Single-Cell Host-Virus Interaction: Correlation Study between Gene Expression Abundance and Viral Load
Time: 7 min
Words: 1.3k words
Updated: 2026-02-27
Reads: 0 times
R
#####################################
# Select Jupyter script execution environment as copyKAT #
#####################################R
library(Seurat)
library(ggplot2)
library(ggpubr)output
Loading required package: SeuratObject
Loading required package: sp
‘SeuratObject’ was built with package ‘Matrix’ 1.6.4 but the current
version is 1.7.0; it is recomended that you reinstall ‘SeuratObject’ as
the ABI for ‘Matrix’ may have changed
Attaching package: ‘SeuratObject’
The following objects are masked from ‘package:base’:
intersect, t
Loading required package: sp
‘SeuratObject’ was built with package ‘Matrix’ 1.6.4 but the current
version is 1.7.0; it is recomended that you reinstall ‘SeuratObject’ as
the ABI for ‘Matrix’ may have changed
Attaching package: ‘SeuratObject’
The following objects are masked from ‘package:base’:
intersect, t
Gene expression data uses "ESCC Paper Reproduction" - "Large Group Reproduction" data as an example
Read workflow RDS data, relative path data/, absolute path /home/mambauser/data/
R
# Workflow rds is data/ProcessID/input.rds
obj = readRDS("data/AY1743044870655/input.rds")
head(obj@meta.data)| orig.ident | nCount_RNA | nFeature_RNA | Sample | mito | raw_Sample | Tissue | Patient | resolution.0.6_d20 | mitorelatedgenes | |
|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <dbl> | <int> | <fct> | <dbl> | <chr> | <fct> | <fct> | <fct> | <dbl> | |
| AAACCTGAGATACACA-1_1 | SeuratProject | 2797 | 1425 | S150T | 4.2903 | GSE145370_S150T | Tumor | S150 | 1 | 3.3249911 |
| AAACCTGAGCTAACTC-1_1 | SeuratProject | 2790 | 1349 | S150T | 1.1470 | GSE145370_S150T | Tumor | S150 | 5 | 1.0035842 |
| AAACCTGAGGAGCGAG-1_1 | SeuratProject | 1768 | 1054 | S150T | 4.7511 | GSE145370_S150T | Tumor | S150 | 1 | 4.0158371 |
| AAACCTGAGGGAAACA-1_1 | SeuratProject | 4455 | 2017 | S150T | 2.2896 | GSE145370_S150T | Tumor | S150 | 8 | 1.8855219 |
| AAACCTGAGTCCCACG-1_1 | SeuratProject | 1422 | 861 | S150T | 0.9845 | GSE145370_S150T | Tumor | S150 | 1 | 0.7032349 |
| AAACCTGAGTGAACAT-1_1 | SeuratProject | 2522 | 1308 | S150T | 1.7843 | GSE145370_S150T | Tumor | S150 | 4 | 1.5463918 |
R
gene_expression_matrix = Seurat::GetAssayData(object = obj, slot = "counts")
gene_expression_matrix[1:3, 1:3]output
Warning message:
“The \`slot\` argument of \`GetAssayData()\` is deprecated as of SeuratObject 5.0.0.
ℹ Please use the \`layer\` argument instead.”
3 x 3 sparse Matrix of class "dgCMatrix"
AAACCTGAGATACACA-1_1 AAACCTGAGCTAACTC-1_1 AAACCTGAGGAGCGAG-1_1
MIR1302-2HG . . .
AL627309.1 . . .
AL627309.2 . . .
“The \`slot\` argument of \`GetAssayData()\` is deprecated as of SeuratObject 5.0.0.
ℹ Please use the \`layer\` argument instead.”
3 x 3 sparse Matrix of class "dgCMatrix"
AAACCTGAGATACACA-1_1 AAACCTGAGCTAACTC-1_1 AAACCTGAGGAGCGAG-1_1
MIR1302-2HG . . .
AL627309.1 . . .
AL627309.2 . . .
Viral gene expression data here uses simulated data based on negative binomial distribution
R
virus_expression = read.delim("sim_virus.matrix")
virus_expression[1:3,1:3]
virus_expression = Matrix::as.matrix(virus_expression)| AAACCTGAGATACACA.1_1 | AAACCTGAGCTAACTC.1_1 | AAACCTGAGGAGCGAG.1_1 | |
|---|---|---|---|
| <int> | <int> | <int> | |
| virus-gene1 | 0 | 1 | 0 |
| virus-gene2 | 0 | 0 | 0 |
| virus-gene3 | 0 | 0 | 0 |
R
# Construct new object, obtain normalized gene expression data
new_obj = CreateSeuratObject(counts = Matrix::rbind2(gene_expression_matrix, virus_expression))
new_obj = NormalizeData(new_obj)
new_obj$Sample = obj$Sampleoutput
Normalizing layer: counts
R
# Obtain viral gene names and host gene names respectively
virus_gene = rownames(new_obj)[grep("^virus", rownames(new_obj))]
host_gene = setdiff(rownames(new_obj), virus_gene)R
# Obtain viral gene expression data
virus_data=GetAssayData(new_obj,assay = "RNA", slot = "data")[virus_gene,]
virus_data_sum = colSums(virus_data)R
# Obtain host gene expression data
host_data = GetAssayData(obj, slot = "data")[host_gene,]
host_gene_num = colSums(host_data != 0)Definition
Gene Expression Abundance: Number of genes expressed in cells
Viral Load: Sum of normalized viral gene expression values (refer to the article above)
R
plot_data=data.frame(virus_load = virus_data_sum,
host_gene_num = host_gene_num,
Sample = new_obj$Sample)
head(plot_data)| virus_load | host_gene_num | Sample | |
|---|---|---|---|
| <dbl> | <int> | <fct> | |
| AAACCTGAGATACACA-1_1 | 2.726585 | 1425 | S150T |
| AAACCTGAGCTAACTC-1_1 | 4.249812 | 1349 | S150T |
| AAACCTGAGGAGCGAG-1_1 | 8.147058 | 1054 | S150T |
| AAACCTGAGGGAAACA-1_1 | 5.170728 | 2017 | S150T |
| AAACCTGAGTCCCACG-1_1 | 13.314534 | 861 | S150T |
| AAACCTGAGTGAACAT-1_1 | 0.000000 | 1308 | S150T |
Plotting
R
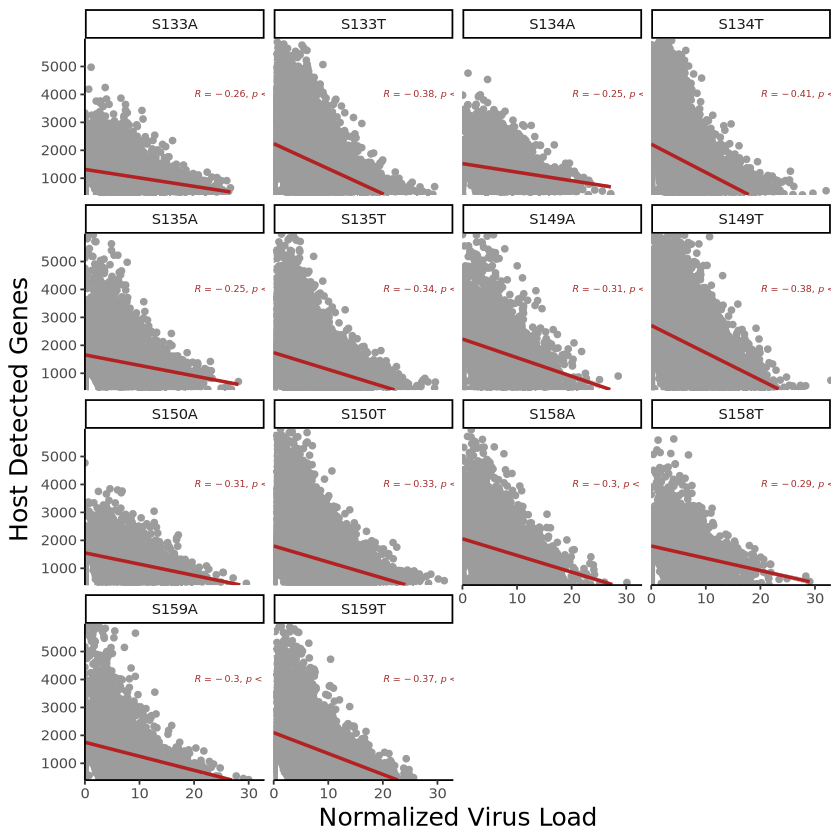
p <- ggplot(plot_data, aes(x = virus_load, y = host_gene_num)) +
geom_point(color = "#9C9C9C", size = 1.5) +
geom_smooth(method = "lm", se = TRUE, color = "Firebrick") + # Add linear fit line
facet_wrap(~Sample) + # Facet by Sample
theme_classic() +
theme(
axis.title = element_text(size = 15), # Increase axis font size
plot.title = element_text(hjust = 0.5, size = 18, face = "bold"), # Center title
) +
labs(
x = "Normalized Virus Load", # x-axis title
y = "Host Detected Genes"# y-axis title
) +
stat_cor(label.x = 20, label.y = 4000, # Position of correlation value
size = 2, # Correlation value font size
color = "brown") + # Add correlation coefficient
scale_x_continuous(expand = c(0,0)) + # Adjust x-axis range
scale_y_continuous(expand = c(0,0),limits = range(plot_data$host_gene_num)) # Adjust y-axis range
poutput
\`geom_smooth()\` using formula = 'y ~ x'
Warning message:
“Removed 152 rows containing missing values or values outside the scale range
(\`geom_smooth()\`).”
Warning message:
“Removed 152 rows containing missing values or values outside the scale range
(\`geom_smooth()\`).”
Save Image
R
ggsave(p, file = "correlation.png", width = 10, height = 10)output
\`geom_smooth()\` using formula = 'y ~ x'
Warning message:
“Removed 152 rows containing missing values or values outside the scale range
(\`geom_smooth()\`).”
Warning message:
“Removed 152 rows containing missing values or values outside the scale range
(\`geom_smooth()\`).”