ATAC + RNA multi-omics cell annotation (RNA expression plus ATAC accessibility)
Document Overview
For multi-omics data from the same cell (simultaneous detection of RNA and ATAC in the same cell), annotation work needs to consider both gene expression (RNA) and chromatin accessibility (ATAC) information sources, improving annotation accuracy and robustness through dual-evidence validation.
Core Concept: Dual-Evidence Integration
RNA Evidence (Transcriptional Output): Identify cell types through expression levels of marker genes, reflecting the current "functional state" of cells.
ATAC Evidence (Regulatory Potential): Identify cell types through chromatin open states of marker gene regions, reflecting the "regulatory state" of cells.
TIP
- In actual cell annotation processes, certain cell types (such as resting neutrophils) may have very low RNA expression, making RNA-only annotation prone to misjudgment; but chromatin structure is intact, and ATAC information can serve as supplementary evidence.
- Sometimes certain cell subpopulations are similar at the transcriptional level but have different regulatory mechanisms. By simultaneously observing RNA and ATAC, we can identify fine categories that are difficult to distinguish with single-omics data.
Dual-Evidence Annotation Strategy
Annotation Workflow
- Step 1: View Clustering Results Visualize clustering results on WNN UMAP
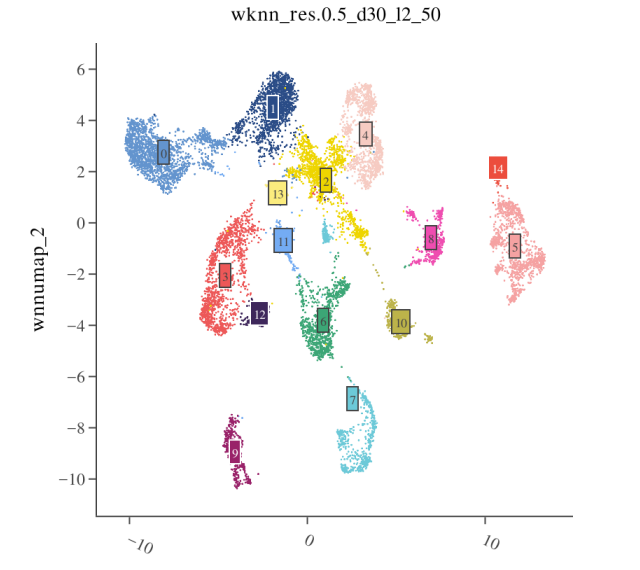
- Step 2: Check RNA Marker Genes Usually before annotation, literature is consulted to determine cell types in the same tissue and their classic Marker gene lists. Functions such as FeaturePlot() or DotPlot() are used to view expression of Marker genes in different cell populations.
# Assuming data is your Seurat object, markers is your Marker gene list of interest
markers <- c("CD3D", "MS4A1", "NKG7", "LYZ", "GNLY", "PPBP") # Example Markers, can be replaced according to actual research
# Visualize expression distribution of each Marker gene in each cluster
DotPlot(
object = data,
features = markers,
dot.scale = 8,
group.by = "seurat_clusters" # This parameter should be filled with wnn joint dimensionality reduction clustering results
)- Step 3: Check ATAC Gene Activity Calculate gene accessibility scores through the GeneActivity() function, and use functions such as FeaturePlot() or DotPlot() to view accessibility of Marker genes in different cell populations.
# Calculate gene activity
gene.activities <- GeneActivity(data)
data[["GeneActivity"]] <- CreateAssayObject(counts = gene.activities)
# Switch to GeneActivity assay
DefaultAssay(data) <- "GeneActivity"
# DotPlot visualization
DotPlot(
object = data,
features = markers,
dot.scale = 8,
group.by = "wnn_clusters" # This parameter should be filled with wnn joint dimensionality reduction clustering results
)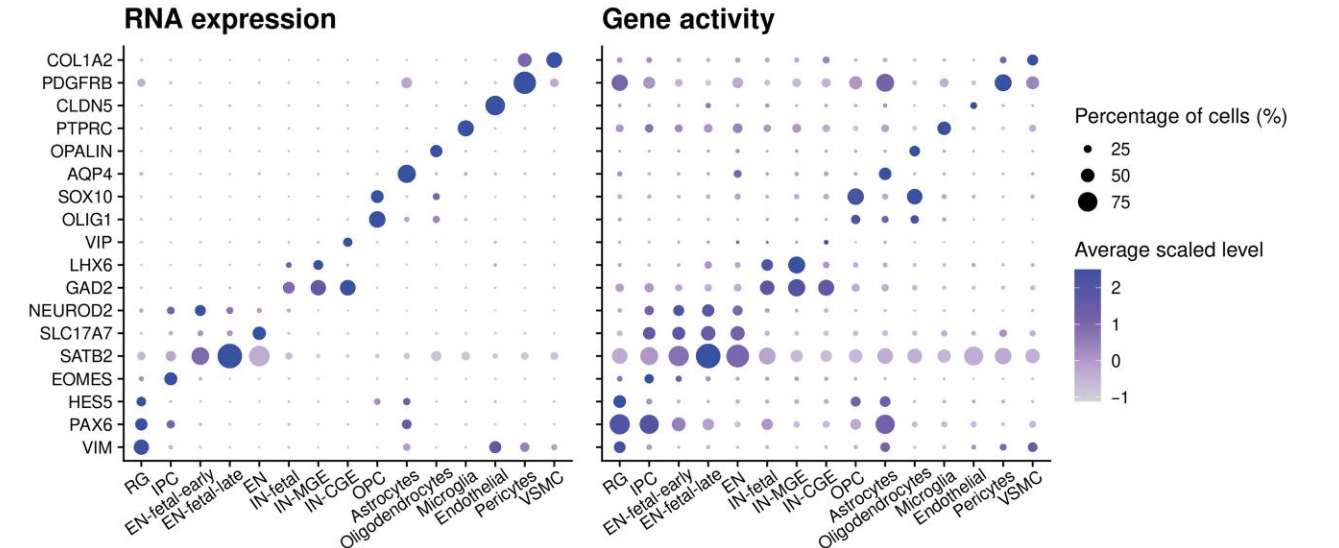
Step 4: Comprehensive Judgment and Annotation
- If RNA and ATAC evidence are consistent, we can be confident that the cluster is a certain cell type.
- If the two are inconsistent, further investigation is needed:
- Data quality issues (unreliable information in one modality).
- Biological reality (such as cells in state transition period).
- Inappropriate clustering resolution (need to adjust resolution and re-cluster).
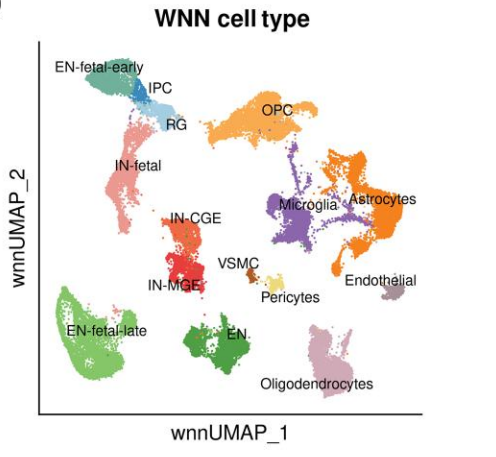
Frequently Asked Questions
Q1: Why do some Marker genes have high expression in RNA but low activity in ATAC?
A: This is a normal phenomenon. Possible reasons include:
- Post-transcriptional Regulation: RNA expression of certain genes may be affected by Post-transcriptional regulation (such as RNA stability) and not completely dependent on chromatin open state.
- Time Delay: Changes in chromatin open state usually precede changes in gene expression. If cells are in a state transition period, ATAC may open first but RNA may not yet be highly expressed (or vice versa).
- Technical Factors: ATAC-seq signals may be affected by factors such as Peak calling and Fragment length distribution, leading to inaccurate activity calculation for certain genes.
- Biological Complexity: Regulation of certain genes may involve distal enhancers. Gene activity calculation only considers gene body and promoter regions, possibly missing important information.
Recommendation: Prioritize Marker genes that show high signals in both RNA and ATAC. These genes have the strongest dual-evidence support and most reliable annotation. For genes with high expression in only a single modality, comprehensive judgment is needed combining other evidence (such as known Marker lists, reference datasets, etc.).
Q2: Is the calculation of gene activity (GeneActivity) accurate? What is its relationship with RNA expression?
A:
Accuracy of Gene Activity:
- Gene activity is calculated based on Peak Fragment counts, affected by factors such as sequencing depth and Peak calling quality, and may have certain errors.
- Gene activity mainly reflects chromatin open state of gene regulatory regions, rather than direct transcriptional activity.
Relationship Between Gene Activity and RNA Expression:
- Positively correlated but not completely consistent: Usually, genes with high gene activity also have higher RNA expression, but the two are not completely corresponding.
- Regulatory time difference: Chromatin opening usually occurs before transcription, so in some cases there may be high ATAC activity but low RNA expression (genes about to be activated).
- Other regulatory mechanisms: RNA expression is also affected by Post-transcriptional regulation, RNA stability, and other factors, not completely dependent on chromatin state.
Recommendation: Use gene activity as supplementary evidence for RNA expression, not as a replacement. When the two are consistent, annotation is more reliable; when they are inconsistent, comprehensive judgment is needed combining other information.
Q3: "Unknown" or undetermined clusters appear in annotation results. What should be done?
A:
Check if it's a technical issue:
- Check quality control indicators of the cluster to confirm if it's low-quality cells or doublets.
- Check the position of the cluster in UMAP space, whether it overlaps or is adjacent to known cell types.
Expand marker gene list:
- Find Top marker genes of the cluster and check if there are tissue-specific or rare cell type markers.
- Use databases (such as CellMarker, PanglaoDB) to find possible cell types.
Temporarily mark as "undetermined":
- If it truly cannot be determined, it can be temporarily marked as "Unknown" or "Unassigned", and these cells should be focused on in subsequent analysis to see if there are new discoveries.
Q4: How to verify the accuracy of annotation results?
A:
Dual-evidence consistency: Check whether marker genes of each annotated cell type are highly expressed in both RNA and ATAC.
UMAP spatial distribution: Clusters of the same cell type should cluster or form continuous regions in UMAP space.
Marker gene specificity: Use DotPlot or VlnPlot to check whether marker genes are specifically highly expressed in target cell types.