ATAC + RNA multi-omics differential accessibility peaks with motif and functional enrichment
Document Overview
Differential peak analysis is a key step in identifying chromatin accessibility differences between different cell populations, crucial for understanding cell type-specific regulatory mechanisms. This document systematically introduces the concept of peaks, methods for calculating differential peaks, motif enrichment analysis, annotation of genes near peaks, and functional enrichment analysis, helping researchers comprehensively interpret chromatin openness differences between cell types or states and their biological significance.
Core Concepts
What are Reads? What is a Fragment? What is a Peak
Before performing differential Peak analysis on scATAC-seq data, we need to clarify several core terms: Reads, Fragments, and Peaks.
- 1) Reads: Raw data directly produced by sequencers, i.e., short DNA sequences. In paired-end sequencing, one DNA/RNA fragment produces one Read from each end, called R1 and R2 respectively.
- 2) Fragment: Original DNA molecules produced by Tn5 transposase cutting in open genomic regions during experiments. In paired-end sequencing, a pair of matched Reads (R1 and R2) can locate a Fragment.
- 3) Peak: On the genome, starting positions of Fragments (i.e., Tn5 cutting sites) densely enrich in open chromatin regions. After statistical analysis, these enriched regions are identified as significant "Peaks".
TIP
- Fragments represent DNA fragments produced by Tn5 enzyme performing a pair of coordinated cuts in open genomic regions, providing direct evidence of open chromatin.
- Peaks can infer biologically functional regions. A Peak represents an open chromatin region, usually associated with regulatory elements (such as promoters, enhancers).
Representation of Peaks
Unlike genes, which have predefined and uniformly named gene names, Peaks are dynamically determined during analysis (such as when using MACS and other tools to Call Peak) based on statistical enrichment signals, so there are no unified names or numbers. Common standard representation methods are as follows, usually annotated in the format of "chromosome-start position-end position".
chr1:9792-10697
chr1:180738-181165
chr1:181194-181660Differential Peak Calculation: Comparison Between Cell Populations
The core purpose of differential Peak calculation is to systematically identify genomic regions with higher or lower openness in specific populations by comparing differences in chromatin open regions (Peaks) between different cell populations (such as different cell types, states, differentiation stages, or disease-related cells).
Methods for Differential Peak Calculation
Taking Signac as an example, Signac uses the same FindMarkers() or FindAllMarkers() functions as RNA differential analysis for differential Peak detection. The following example calculates differential Peaks for each cell population compared to other populations.
# First set ATAC assay as default
DefaultAssay(obj) <- "ATAC"
# Using seurat_cluster clustering results as an example
diff_peak_res <- FindAllMarkers(
object = obj, # Assuming we have already normalized ATAC data for obj
group.by = "seurat_cluster", # Specify cell clustering results, if not specified, defaults to Ident(obj) clustering results
min.pct = 0.1, # Detected in at least 10% of cells
logfc.threshold = 0.25, # Minimum log2 fold change
only.pos = TRUE, # Only retain Peaks higher in target population
test.use = "wilcox" # Wilcoxon rank-sum test (default)
)TIP
FindAllMarkers()is suitable for automatically comparing each cell population with all other populations at once, very suitable for comprehensive differential analysis of multi-component clustering (such as multiple cell types or states).FindMarkers()is suitable for direct comparison between two groups (such as disease group vs. control group), enabling flexible one-to-one differential Peak screening, more convenient for precise comparison between specific groups.
Results of Differential Peak Calculation
The table below shows the results of differential Peak analysis for each cell population using the FindAllMarkers() function from the Signac package. Each row corresponds to a specific chromatin open Peak. Through these fields, we can systematically compare openness levels between cell populations in different genomic regions, providing a data foundation for downstream functional annotation and regulatory mechanism exploration.
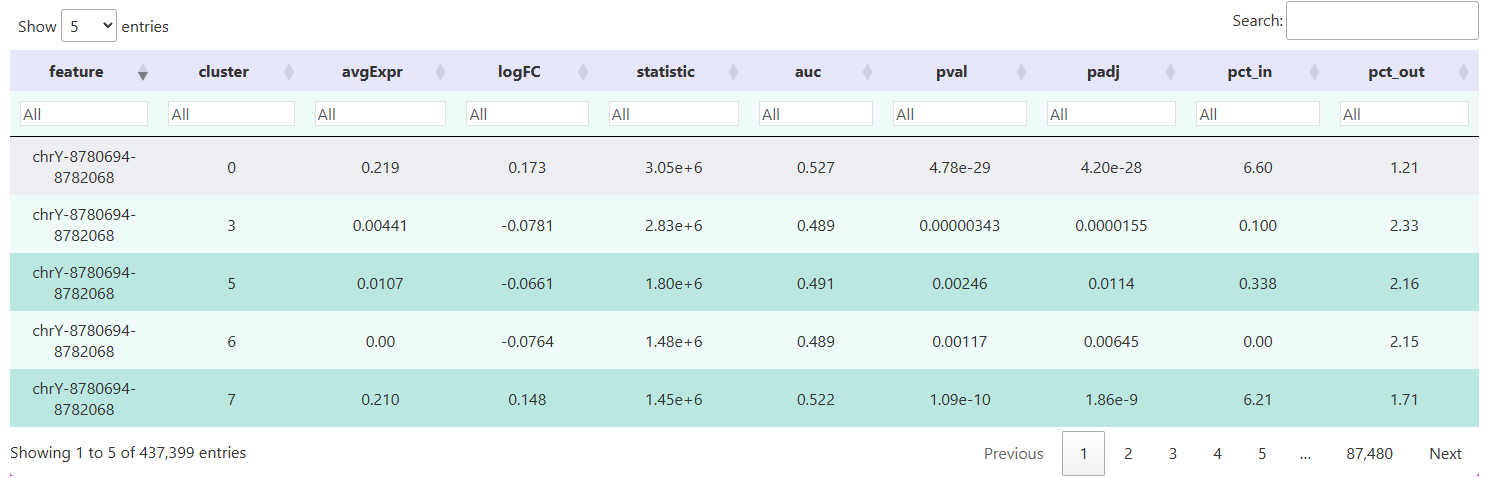
feature: Unique identifier for the differential Peak.cluster: Corresponding cell category or clustering label, showing the cell population where each Peak is most significantly open.p_val: Raw P-value from statistical testing, measuring the significance of openness differences between two cell groups at this Peak.p_val_adj: Adjusted P-value (usually using Benjamini-Hochberg method for FDR correction).avg_log2FC: Average log fold change, reflecting the fold change of Peak openness in the target population relative to the control population (log base 2).pct.1: Proportion of cells in the target population where the Peak is detected.pct.2: Proportion of cells in the control population where the Peak is detected.
TIP
Filtering Criteria:
- Significance:
p_val_adj < 0.05(or stricter, such as 0.01). - Effect size:
avg_log2FC > 0.25(or higher, such as 0.5), indicating openness in the target population is at least 1.2 times that of the control population. - Detection rate:
pct.1 > pct.2, ensuring the Peak has a higher detection rate in the target population.
Display of Differential Peaks
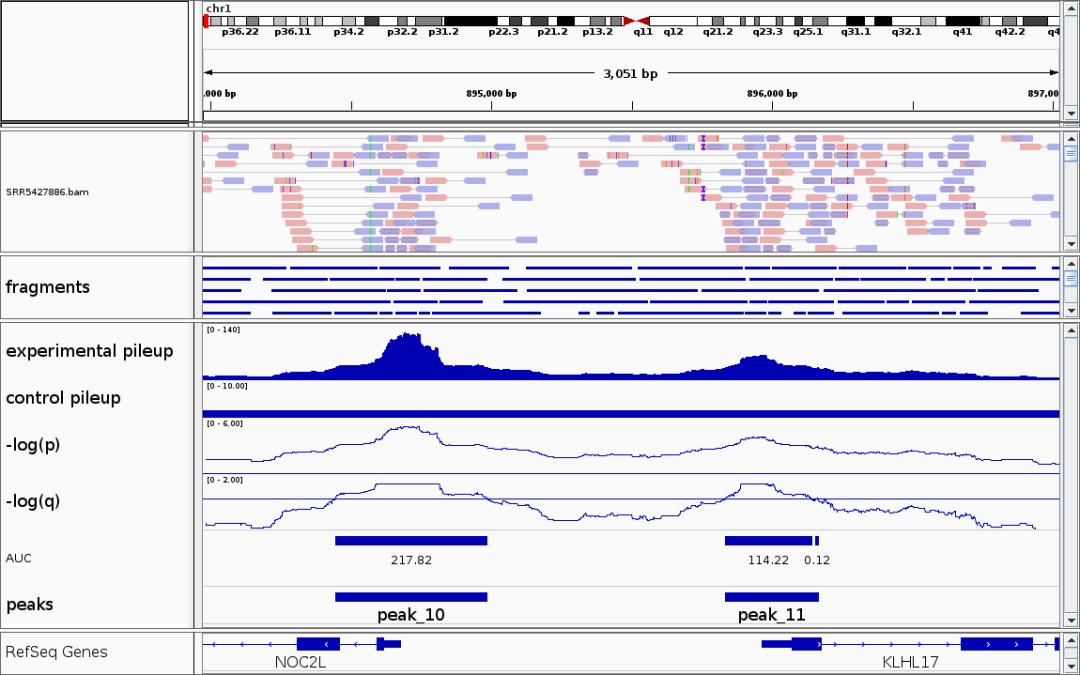
The following shows two Peaks specifically open in the cluster2 cell population. Differential Peaks represent chromatin that is more open in some cell populations, suggesting these regions may be related to cell function and gene regulation.
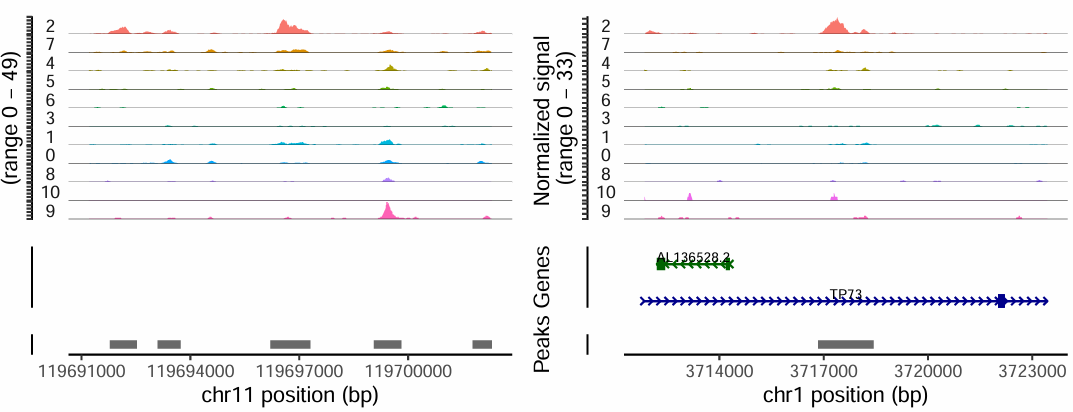
TIP
- Genomic regions of Marker genes also usually have cell population-specific chromatin openness.
- If a gene is a Marker for a certain cell type, Peaks near its promoter or enhancer generally show specific openness in that cell population.
- We can assist cell type annotation by visualizing chromatin accessibility of Marker genes.
Motif Enrichment Analysis of Differential Peaks
Definition of Motif
Transcription factors need to bind to DNA open regions (Peaks) to exert regulatory effects. Each transcription factor usually has preferred binding sequences (e.g., SP1 prefers GC-rich sequences). These sequence patterns are Motifs. Therefore, Motif (sequence motif) can be understood as the DNA sequence pattern of transcription factor binding sites, usually composed of 6-20 base pairs. As shown below:

Purpose of Motif Enrichment Analysis
- Motif analysis aims to infer key transcription factors involved in regulating these peaks by identifying specific motifs enriched in differentially open peaks. This helps reveal core regulatory molecules between cell subpopulations and further resolve cell type or state-specific transcriptional regulatory networks and functional mechanisms.
Implementation of Motif Enrichment Analysis
Taking Signac as an example, Signac uses the AddMotifs() function to add motif information to Seurat objects and uses FindMotifs() for motif enrichment analysis. Below are key code examples (using human, hg38 as an example):
library(Signac)
library(JASPAR2022)
library(TFBSTools)
# Load motif database (using human species as an example)
pfm <- getMatrixSet(
x = JASPAR2020,
opts = list(collection = "CORE", tax_group = "vertebrates", all_versions = FALSE)
)
# Add motif information to ATAC object (e.g., atac)
DefaultAssay(obj) <- "ATAC"
obj <- AddMotifs(
object = obj, # Your Seurat/Signac object
genome = BSgenome.Hsapiens.UCSC.hg38, # Note: load appropriate genome package
pfm = pfm
)
# Perform motif enrichment analysis on peaks of interest, using all peaks in obj as background peaks
motif.results <- FindMotifs(
object = obj,
features = diff_peak # For example, list of differential peaks from differential analysis
)
head(motif.results)| motif | observed | background | pct.obs | pct.bkg | fold.enrich | pvalue | motif.name | p.adjust |
|---|---|---|---|---|---|---|---|---|
| MA0497.1 | 556 | 8315 | 49.16 | 20.79 | 2.365 | 0 | MEF2C | 0 |
| MA0052.4 | 533 | 7948 | 47.13 | 19.87 | 2.372 | 0 | MEF2A | 0 |
| MA0773.1 | 398 | 4930 | 35.19 | 12.33 | 2.855 | 0 | MEF2D | 0 |
| MA0660.1 | 345 | 4002 | 30.50 | 10.01 | 3.049 | 0 | MEF2B | 0 |
| MA1151.1 | 286 | 3206 | 25.29 | 8.02 | 3.155 | 0 | RORC | 0 |
| MA0592.3 | 350 | 4669 | 30.95 | 11.67 | 2.651 | 0 | MEF2A | 0 |
motif: The Motif ID in the database, usually representing a specific transcription factor's binding sequence.observed: The number of target Peaks (such as differential Peaks) containing this Motif.background: The number of background Peaks (control group or all Peaks) containing this Motif.pct.obs: The percentage of target Peaks with this Motif (= observed/total target Peaks × 100%).pct.bkg: The percentage of background Peaks with this Motif (= background/total background Peaks × 100%).fold.enrich: The enrichment fold of the Motif in target Peaks, = pct.obs / pct.bkg. Larger values indicate greater Motif enrichment in differential Peaks.pvalue: The raw P-value for statistical significance of Motif Enrichment Analysis. Smaller values mean enrichment is less likely to be random.motif.name: The transcription factor name corresponding to the Motif, facilitating intuitive identification of regulatory factors.p.adjust: The P-value after multiple hypothesis testing correction (such as FDR correction). p.adjust < 0.05 is often considered significant enrichment.
Through the above indicators, we can determine which Motifs (and their corresponding transcription factors) are significantly and strongly enriched in differential Peaks, thereby revealing key TFs that may be involved in regulation.
Background Peaks:
- Refers to a set of Peaks used as controls in Motif Enrichment Analysis, usually representing all Peaks or Peaks with no significant differences. Their role is to provide a baseline for the frequency of a certain Motif in Peaks of interest (such as differential Peaks).
- By comparing the enrichment level of a Motif in differential Peaks versus background Peaks, we can determine whether the Motif is significantly enriched under specific biological states.
- The FindMotif() function can provide a feature vector as a background set through the background parameter, or provide a number to specify the number of randomly selected features as a background set.
TIP
For detailed content on Motif Analysis of scRNA-seq+scATAC-seq multi-omics data, please refer to "Single-cell ATAC_RNA Multi-omics Motif Analysis".
Gene Annotation Near Differential Peaks
Principle of Peak-Gene Association (Gene Annotation)
- Peaks themselves are genomic coordinates and need to be associated with nearby genes through annotation to understand their biological functions.
- Usually, Peaks are associated with the nearest gene (regardless of distance).
Gene Annotation Methods
Taking Signac method as an example:
# If object obj does not yet have annotation information, it needs to be added first
# Assuming relevant packages are already loaded (Signac, EnsDb, GenomicRanges, etc.)
# Add annotation information (e.g., for human species data, skip if already added)
DefaultAssay(obj) <- "ATAC"
annotation <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
# Set gene chromosome names to be consistent with obj
seqlevelsStyle(annotation) <- "UCSC"
genome(annotation) <- "hg38"
Annotation(obj) <- annotation
# Find the nearest gene for each differential Peak
closest_genes <- ClosestFeature(
object = obj,
regions = diff_peak # Or custom Peak regions
)
head(closest_genes)Actual output is as follows:
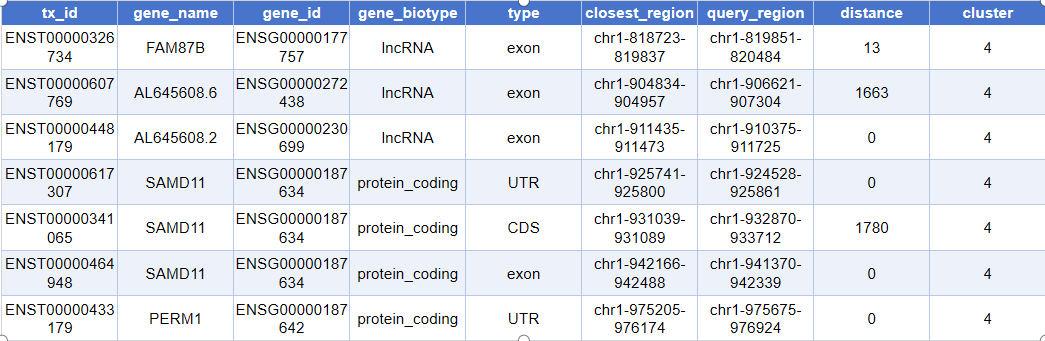
tx_id: Unique ID for transcript exons/fragments (such as ENSE numbers)gene_name: Symbol name of the gene (such as TP53, GAPDH, etc.)gene_id: Gene ID in the database (such as ENSG-prefixed numbers)gene_biotype: Gene type (e.g., lincRNA, protein_coding, pseudogene, etc.)type: Specific region type annotated (such as exon, promoter, etc.)closest_region: Genomic segment closest to the peak (its coordinate interval)query_region: Genomic position information of the annotated peak (coordinate interval)distance: Distance from peak to the nearest end of the corresponding genomic segment (in base pairs bp)
Functional Enrichment Analysis of Differential Peaks
We have completed gene annotation of peaks through the above sections. Next, we perform functional enrichment on these genes using databases such as GO and KEGG. The analysis approach and workflow are similar to conventional differential gene functional enrichment.
By performing functional enrichment on genes annotated from differential peaks, we can systematically reveal the main biological processes and signaling pathways involved by these genes, thereby providing preliminary basis for explaining the potential biological significance of differential peaks.
TIP
Functional enrichment analysis of differential peaks essentially performs enrichment and functional annotation analysis on neighboring genes corresponding to these peaks.
Common Functional Enrichment Databases
GO (Gene Ontology) Enrichment:
- Biological Process (BP): Describes biological processes genes participate in, such as "cell proliferation", "immune response".
- Molecular Function (MF): Describes molecular functions of gene products, such as "DNA binding", "kinase activity".
- Cellular Component (CC): Describes cellular locations where gene products are located, such as "nucleus", "mitochondria".
KEGG Pathway Enrichment:
- KEGG (Kyoto Encyclopedia of Genes and Genomes) provides annotations for signaling pathways, metabolic pathways, etc.
- Can help understand which key biological pathways are affected by differential peaks.
Other Databases:
- Reactome: Provides detailed pathway annotations.
- MSigDB: Contains various gene sets, such as Hallmark gene sets, cell type-specific gene sets, etc.
Functional Enrichment Analysis Examples
The figure below shows visualization results of GO functional enrichment analysis on genes associated with differential peaks. The vertical axis displays significantly enriched GO terms, helping to quickly identify representative functional categories and their significance levels, thereby providing reference for understanding biological functions that differential peaks may involve.
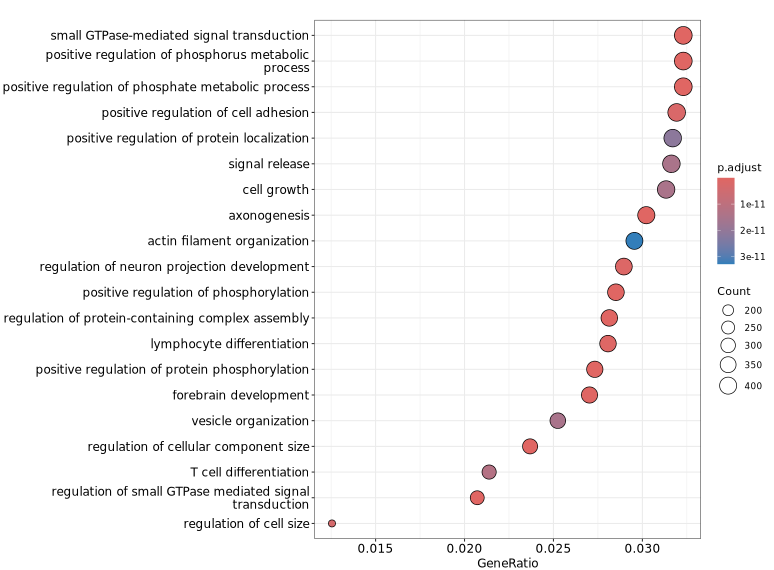
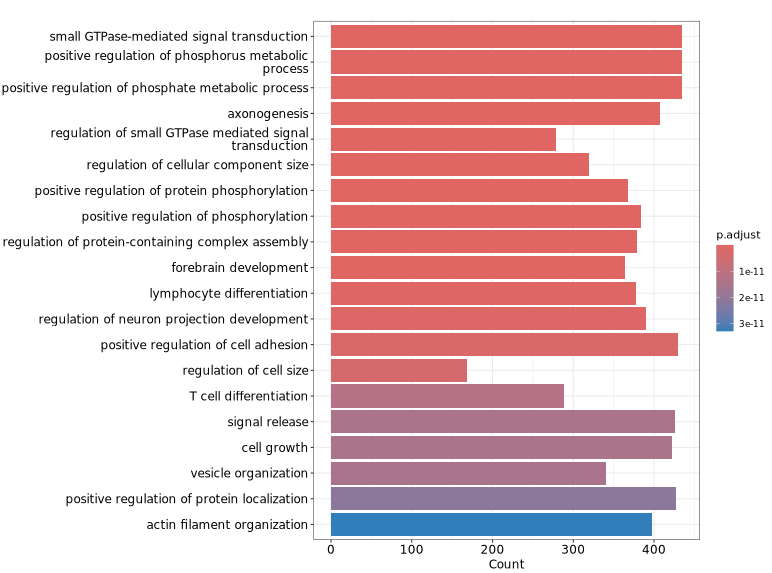
GeneRatio / BgRatio: Proportion of genes in the target gene set containing this function/pathway vs. proportion in the background gene set.p.adjust: Significance of enrichment. p.adjust < 0.05 is usually considered significant enrichment.Count: Number of genes in the target gene set belonging to this function/pathway.
TIP
Term: In functional enrichment analysis, "term" usually refers to a functional category in gene set databases (such as GO, KEGG), for example, a specific biological process (such as "apoptosis"), molecular function (such as "DNA binding"), or pathway (such as "Wnt signaling pathway").
Frequently Asked Questions
Q1: What are the purposes and applications of differential peak analysis?
A: The main purpose of differential peak analysis between cell populations is to identify which chromatin regions (peaks) have differences in openness between different cell populations. This can help us:
- Discover population-specific regulatory elements
- Reveal which regulatory regions such as enhancers and promoters are more open in certain cell types, more likely regulating cell-specific functions.
- Infer related genes and their biological functions
- By associating differential peaks with nearby genes, infer that expression of these genes may be affected by these regulatory regions, and further perform functional enrichment analysis (such as GO, KEGG).
- Screen potential regulatory factors
- Can combine motif enrichment and other analyses to predict key transcription factors active in specific cell populations.
Q2: In motif enrichment analysis results, how to determine which transcription factors are most important?
A: Can comprehensively consider the following aspects:
Prerequisite selection: When performing motif enrichment analysis, it is recommended to prioritize selecting differential peaks with more significant differences (such as peaks with smaller p-values or higher fold-changes) as the input set for enrichment analysis, which can improve the biological relevance and reliability of the analysis.
Indicator judgment:
- Fold enrichment: Larger values indicate stronger enrichment of the TF's motif in top peaks.
- Significance (p.adjust): TFs with
p.adjust < 0.05(or stricter) are more credible. - Biological knowledge: Combined with research background, prioritize TFs known to be important in this cell type or process.
- Consistency: If multiple related motifs all enrich the same TF, it indicates the TF is more important.