ATAC + RNA multi-omics QC guide and key metrics
Purpose of Quality Control
Remove abnormal barcodes such as cell debris, doublets, and dead cells to ensure analysis only includes high-quality, genuine single cells.
Correct State of Oil Droplet Encapsulating Single Cell
The figure below shows the ideal situation: one oil droplet only encapsulates one complete cell. However, in actual experiments, oil droplets may also encapsulate multiple cells (doublets), or oil droplets may contain free cell debris. Both should be removed during quality control.

Key Indicators and Recommendations
RNA Quality Control
Core indicators (thresholds need to be flexibly adjusted according to data):
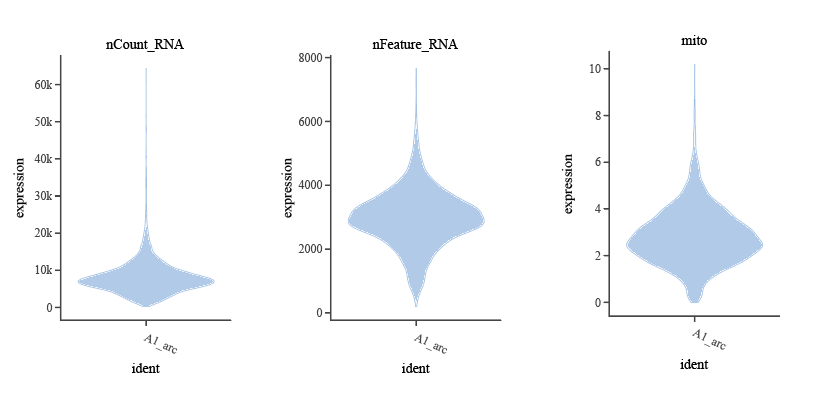
- Number of genes detected per cell (
nFeature_RNA): Too low suggests cell debris rather than real cells, too high suggests doublets, not independent single cells. Common threshold recommendation: 200~6000 (specific thresholds can be appropriately adjusted in combination with violin plots). - Total UMI per cell (
nCount_RNA): Reflects the total capture of all transcript molecules in each cell, usually changes in coordination with gene number. Common filtering range: 500–10,000 (specific thresholds can be appropriately adjusted in combination with violin plots). - Mitochondrial gene percentage (
percent.mt): This indicator is used to filter possible dead cells. Mitochondrial percentage above 20% or 25% often indicates cells are stressed, damaged, or in a dying state. Common recommended threshold: ≤10%~20%.
# Calculate mitochondrial gene percentage (using human genes as an example, mitochondrial genes usually start with "MT-")
data[["percent.mt"]] <- PercentageFeatureSet(
object = data,
pattern = "^MT-"
)# Plot RNA quality control violin plots (nFeature_RNA, nCount_RNA, percent.mt)
VlnPlot(
object = data,
features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3,
pt.size = 0
)Outliers in violin plots (such as extremely low or high nFeature_RNA, abnormally elevated percent.mt, etc.) often reflect poor-quality cells, and these regions are suitable as filtering thresholds. It is recommended to flexibly fine-tune thresholds in combination with empirically recommended ranges and violin plot morphology to maximize retention of high-quality cells and remove noise.
ATAC Quality Control
It is recommended to evaluate the following indicators:
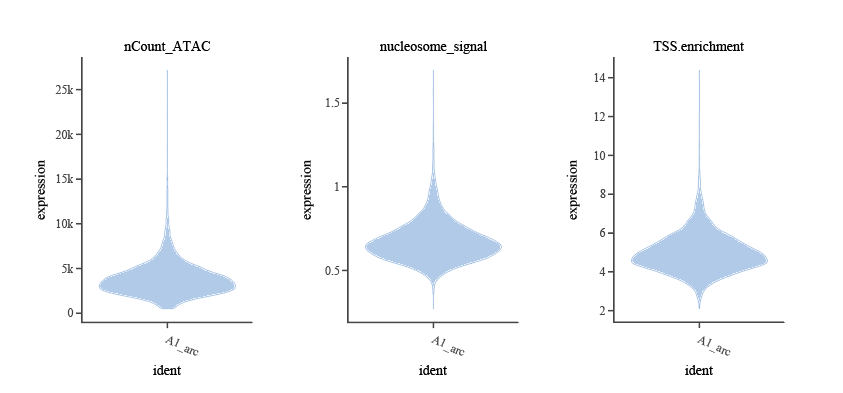
- TSS enrichment score (
TSS.enrichment): An indicator measuring cell chromatin state. High TSS enrichment suggests good chromatin open state, usually corresponding to good cell survival and transcriptional homeostasis. Low TSS enrichment scores are common in chromatin degradation, cell death, or technical noise. Can be calculated usingTSSEnrichment(). Common threshold recommendation: ≥1–2 (recommended lower limit, specific thresholds need to be appropriately adjusted in combination with violin plots).
# Run TSS enrichment score calculation
data <- TSSEnrichment(
object = data,
assay = "ATAC"
)
# Results will be added to the TSS.enrichment column in meta.data- Nucleosome signal (
nucleosome_signal): Describes the ratio of Mononucleosome Fragments (~147bp) to Nucleosome-free Fragments (<100bp). High-quality cells, due to intact chromatin structure, will show periodic Peaks in Fragment length distribution with alternating high and low, approximately 200bp multiples, reflecting the physiological state of nucleosome arrangement and chromatin packaging within cells. Abnormally elevated nucleosome signals suggest non-physiological states such as cell apoptosis, degradation, or excessive doublets. Common recommended threshold: ≤2, values greater than this are recommended for removal.
# Calculate nucleosome signal (nucleosome_signal)
data <- NucleosomeSignal(
object = data,
assay = "ATAC"
)
# Results will be added to the nucleosome_signal column in meta.data- Total ATAC Fragments (
nCount_ATAC): This indicator is similar to the abovenCount_RNA, filtering debris or doublets from the ATAC level. Common recommended range: 1,000–80,000 (specific thresholds need to be appropriately adjusted in combination with violin plots).
# Plot ATAC quality control violin plots (TSS.enrichment, nucleosome_signal, nCount_ATAC)
VlnPlot(
object = data,
features = c("TSS.enrichment", "nucleosome_signal", "nCount_ATAC"),
ncol = 3,
pt.size = 0
)Similarly, use violin plots to help us intuitively visualize the distribution of various QC indicators at the ATAC level, assist in determining reasonable filtering thresholds, and identify abnormal or low-quality cells.
Frequently Asked Questions
Q1: I used the Signac official tutorial to process SeekArc™ Tools data, and the tutorial uses a singlecell.csv file, but you didn't provide this file to me?A: The tutorial you're using is probably for processing scATAC-seq single-omics data. In single-omics tutorials, the singlecell.csv file is used to calculate the pct_reads_in_peaks indicator for cell filtering. Signac provides a specific tutorial for single-cell multi-omics: https://stuartlab.org/signac/articles/pbmc_multiomic
Q2: Why is the pct_reads_in_peaks indicator not used to filter cells in Signac multi-omics (ATAC + RNA) analysis?A: pct_reads_in_peaks (percentage of Reads in Peaks) is mainly used for single-omics ATAC quality control, reflecting signal-to-noise ratio; in single-omics, there is no other data support, so it needs to be strictly used to remove low-quality cells. However, in multi-omics data, RNA information can serve as a complement to ATAC data quality control. If a cell's RNA data performs well, even if pct_reads_in_peaks is slightly low, it may still be a biologically real and valuable cell. Some cell subtypes naturally have low pct_reads_in_peaks, and forced filtering may lose key information. Therefore, Signac focuses more on robust indicators such as TSS.enrichment and nucleosome_signal in multi-omics, cross-validating overall quality through ATAC and RNA, without requiring mandatory filtering using pct_reads_in_peaks.
Q3: How do neutrophils affect quality control indicators in multi-omics data?A:
- When neutrophils are activated (such as by inflammatory stimulation), chromatin will depolymerize and be released extracellularly, forming "Neutrophil Extracellular Traps" (NETs) bound to histones to clear pathogens. Such events produce large amounts of randomly distributed small DNA Fragments, which manifest in sequencing data as:
- Significant decreases in indicators such as TSS enrichment and Overlap Peaks;
- These fragments can account for up to 60% on chromosomes, and most are difficult to be assigned to Peak regions called by ATAC;
- Samples containing large numbers of neutrophils in this state are not suitable for scATAC-seq sequencing.
- Unactivated neutrophils in resting (terminally differentiated) state, although having relatively low overall transcriptional activity, have intact chromatin structure. Various conventional quality control indicators (such as TSS.enrichment, Overlap Peaks, etc.) are usually within normal ranges with only relatively lower median values for gene expression (nFeature_RNA, nCount_RNA) and total ATAC fragments (nCount_ATAC). This is a physiological phenomenon and is also common in actual data.