单细胞 scATAC-seq & scRNA-seq 双组学标准分析:降维与聚类
概述
与单组学分析不同,单细胞多组学数据的降维聚类有RNA降维聚类 、 ATAC降维聚类和联合RNA和ATAC进行加权最近邻(Weighted Nearest Neighbors, WNN)降维聚类 共三种降维聚类的策略。
一、为什么需要分别降维和联合降维?
单细胞多组学数据的降维策略包含三个层次:
RNA 单独降维聚类:利用基因表达信息识别转录活跃的细胞类型,反映细胞的“功能状态”(转录输出)。
ATAC 单独降维聚类:基于染色质可及性模式识别细胞类型,反映细胞的“调控潜能”(调控状态)。
联合降维(WNN):整合 RNA 和 ATAC 两种信息源,构建加权近邻图,实现更稳健、更高分辨率的细胞类型识别。
二、RNA 端降维与聚类
1) 数据标准化与特征提取
SCTransform 标准化:消除测序深度、线粒体基因占比等技术因素的影响,保留生物学变异。
主成分分析(PCA):将高维基因表达数据降维到低维空间(通常是前
30个主成分),保留主要变异信息。
2) UMAP 可视化与聚类
UMAP 降维:将高维数据可视化到
2D空间,便于直观观察细胞分布和聚类结果。聚类分析:在
pca空间构建 K 近邻图(KNN),使用 FindNeighbors()和FindClusters()函数进行聚类。
library(Seurat)
# SCTransform标准化(矫正线粒体基因影响)
data <- SCTransform(data, assay = "RNA", vars.to.regress = "percent.mt", verbose = FALSE)
# PCA降维
data <- RunPCA(data, assay = "SCT", npcs = 30, verbose = FALSE)
# UMAP降维(也可`RunTSNE()`进行TSNE降维)
data <- RunUMAP(data, reduction = "pca", dims = 1:30, reduction.name = "umap")
# 聚类分析
data <- FindNeighbors(data, reduction = "pca", dims = 1:30)
data <- FindClusters(data, resolution = 0.6)
# 可视化
DimPlot(data, reduction = "harmony.rna.umap", label = TRUE)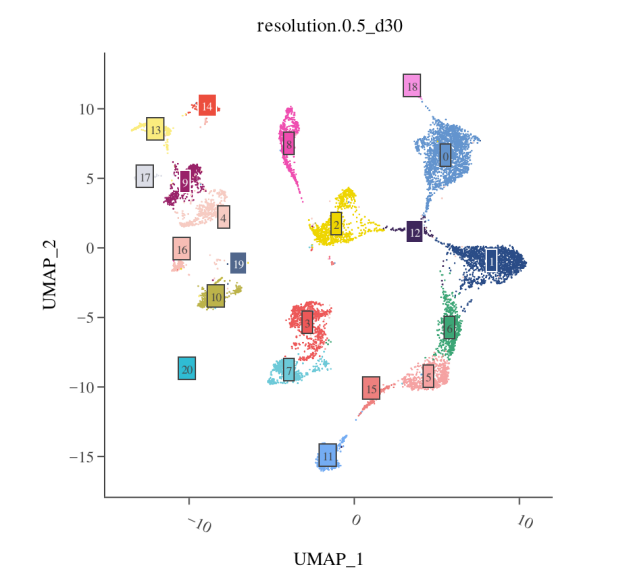
三、ATAC 端降维与聚类
1) TF-IDF 标准化与特征选择
TF-IDF 变换:scATAC-seq 数据是稀疏的计数矩阵。需要使用 RunTFIDF() 函数对每个细胞的 peak 计数进行标准化。此方法能减弱那些在多数细胞中开放(缺乏特异性信息)的 peak 的影响,另一方面也有助于缓解因测序深度差异带来的批次偏差。
Top Features 筛选:使用
FindTopFeatures()函数,通常选择最低分位数(如min.cutoff = 'q0')以上的 peak,可以减少计算负担并提高降维质量,也可以显著提升后续 LSI 的计算效率。。
2) LSI 降维
- 潜在语义索引(LSI):类似于 PCA,将高维 peak 矩阵降维到低维空间(通常 50 个 LSI 维度)。
TIP
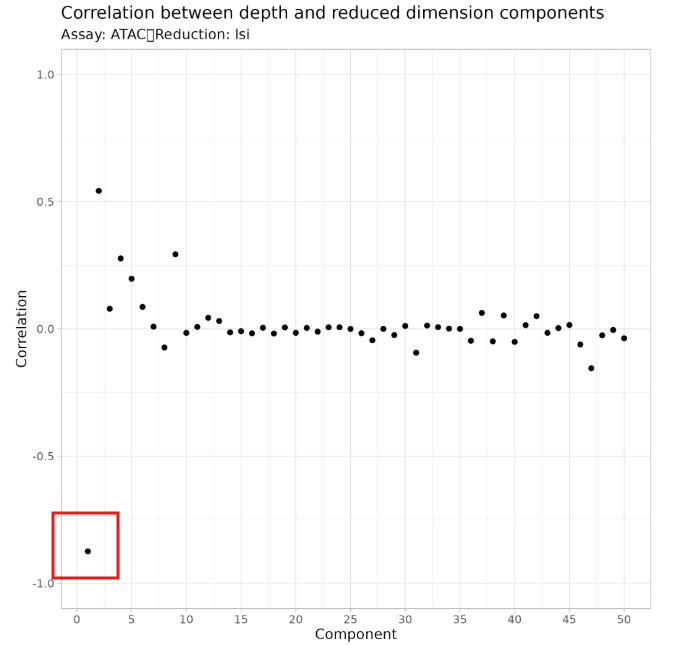
- LSI_1 主要捕获测序深度的差异,而非细胞类型差异。包含 LSI_1 会导致聚类结果受测序深度主导,而非真正的生物学变异。
- 从 LSI_2 开始使用,可以更准确地反映染色质状态的生物学差异。
3) UMAP 可视化与聚类
UMAP 降维:将高维数据可视化到
2D空间,便于直观观察细胞分布和聚类结果。聚类分析:在
lsi空间构建 K 近邻图(KNN),使用 FindNeighbors()和FindClusters()函数进行聚类。
library(Signac)
# ATAC端TF-IDF变换与特征筛选
data <- RunTFIDF(data, assay = "ATAC")
data <- FindTopFeatures(data, assay = "ATAC", min.cutoff = 'q0')
# LSI降维
data <- RunSVD(data, assay = "ATAC", reduction.key = "LSI_", n = 50)
# UMAP降维(也可`RunTSNE()`进行TSNE降维)
data <- RunUMAP(data, reduction = "lsi", dims = 2:50, reduction.name = "umap.atac")
#聚类分析
data <- FindNeighbors(data, reduction = "lsi", dims = 2:50)
data <- FindClusters(data, resolution = 0.6)
# ATAC端可视化
DimPlot(data, reduction = "umap.atac", label = TRUE)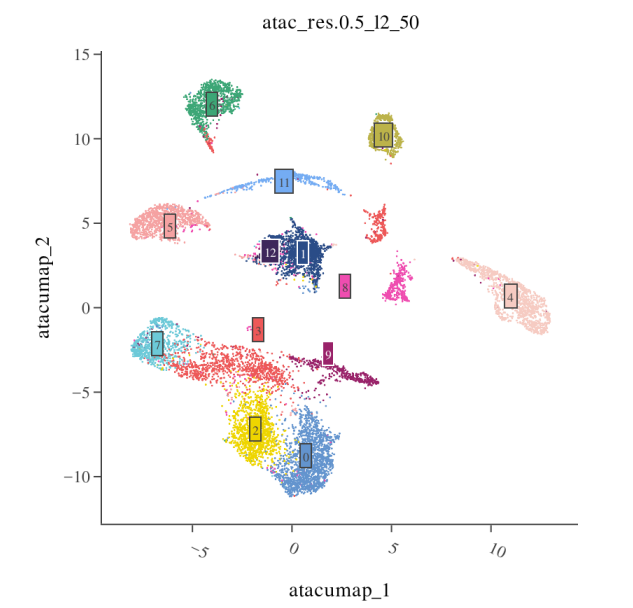
四、联合降维:WNN(Weighted Nearest Neighbors)
1) WNN 原理
加权最近邻的核心思想:对于每个细胞,WNN 会同时考虑其在 RNA 维度和 ATAC 维度中的近邻关系,并根据两种模态的信息量动态调整权重。
权重计算机制:如果某个细胞在 RNA 维度中的信息丰富(与其他细胞区分度高),RNA 的权重会更高;反之,如果 ATAC 维度的信息更可靠,ATAC 的权重会更高。自适应权重使得 WNN 能够充分利用两种模态的优势,同时避免弱模态的干扰。
2) WNN 实现步骤
- 第一步:构建加权近邻图
obj <- FindMultiModalNeighbors(
obj,
reduction.list = list("pca", "lsi"),
dims.list = list(1:30, 2:50)
)参数说明:
reduction.list:指定用于构建近邻图的降维结果列表(RNA 和 ATAC)。dims.list:分别指定 RNA 和 ATAC 使用的维度范围。注意 ATAC 从第 2 维开始。第二步:在 WNN 空间进行 UMAP 降维
obj <- RunUMAP(
obj,
nn.name = "weighted.nn",
reduction.name = "wnn.umap",
reduction.key = "wnnUMAP_"
)参数说明:
nn.name:指定使用哪个近邻图,这里使用weighted.nn。reduction.name:保存降维结果的名字,用于后续可视化。第三步:基于 WNN 图进行聚类
obj <- FindClusters(
obj,
graph.name = "wnn",
resolution = 0.4,
algorithm = 3 # 3 代表 Leiden 算法
)参数说明:
graph.name:指定使用wnn图(加权共享最近邻图)进行聚类。resolution:聚类分辨率,数值越大,得到的聚类数越多。建议从 0.2 开始尝试。algorithm:聚类算法,3代表 Leiden 算法(推荐)。
3) WNN 结果的可视化与验证
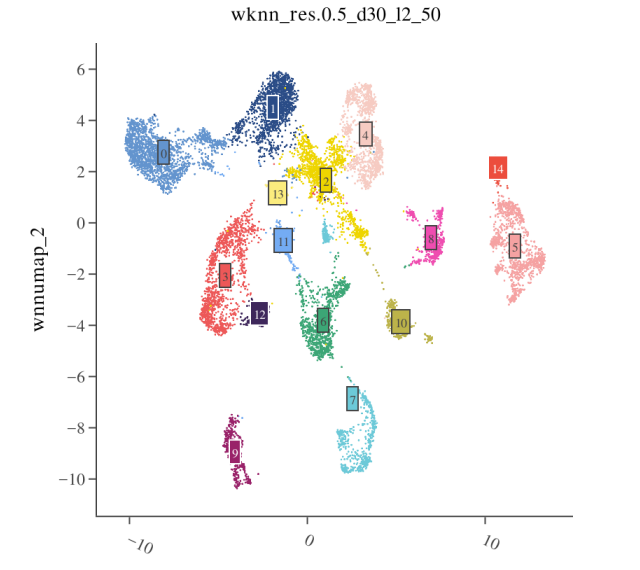
五、常见问题
Q1:为什么 ATAC 的 LSI 降维要从第 2 个维度开始,而不是第 1 个?
A:LSI 的第一个维度(LSI_1)主要捕获测序深度的差异,而非生物学变异。这与 PCA 不同,PCA 的第一主成分通常包含最多的生物学信息。在 ATAC-seq 数据中,测序深度差异很大(从数千到数万片段),LSI_1 会反映这种技术变异,而非细胞类型的真实差异。如果使用 LSI_1,聚类结果可能会被测序深度主导,导致高深度细胞聚集在一起,低深度细胞聚集在一起,而非按照细胞类型聚类。因此,从 LSI_2 开始使用可以更准确地捕获生物学变异。
Q2:RNA 和 ATAC 单独聚类的结果不一致,这是正常的吗?
A:是的,这是正常且常见的现象。RNA 和 ATAC 两种模态捕获的是不同类型的信息:
- RNA 反映基因的转录输出,受转录活性、RNA 稳定性等因素影响。
- ATAC 反映染色质的可及性,受转录因子结合、表观遗传修饰等因素影响。
因此,两种模态的聚类结果可能存在差异:
- 如果差异较小,说明两种模态信息一致,细胞类型在转录和调控层面都比较清晰。
- 如果差异较大,可能提示:
- 某些细胞亚群在转录水平相似但调控状态不同(或反之),这正是联合降维能够解决的场景。
- 数据质量问题,某一模态的信息不够可靠。
- 生物学真实情况,如某些细胞处于状态转换期,转录和调控不同步。
建议同时查看 RNA、ATAC 和 WNN 三种聚类结果,WNN 的结果通常是最稳健的。
Q3:如何验证 WNN 联合降维的结果是否合理?
A:可以从以下几个方面验证:
与已知 marker 基因的一致性:检查已知的细胞类型 marker 基因(如 CD3D 标记 T 细胞、MS4A1 标记 B 细胞)是否在对应的 WNN 聚类中高表达。
RNA 和 ATAC 的一致性:对比 RNA、ATAC 和 WNN 三种聚类结果,观察 WNN 是否合理整合了两种模态的信息。如果 WNN 聚类与 RNA、ATAC 聚类都有一定的对应关系,说明整合是合理的。
生物学合理性:检查聚类结果是否符合生物学预期。例如,发育相关的细胞类型应该在 UMAP 空间中相邻;功能相似的细胞类型应该聚集在一起。