单细胞 scATAC-seq & scRNA-seq 双组学标准分析:Peak2Gene 分析
文档概览
Peak2Gene(Peak-Gene linking)分析是一种专为单细胞多组学(ATAC+RNA)数据设计的方法。其核心目标是识别基因表达与其附近染色质可及性峰(peaks)之间的显著调控关系。该方法通过统计每个细胞中基因表达量与其附近 peaks 的 ATAC 信号强度间的相关性,并利用广义线性模型校正 GC 含量、peak 长度、距离等技术偏置,从而推断哪个 peak 可能参与调控哪些基因。Peak2Gene 分析不仅帮助揭示顺式调控网络(如增强子-基因连接),还能与 motif 富集/活性结果结合,系统性挖掘关键转录因子的直接靶基因和调控作用,是多组学调控机制研究的基础工具之一。
一、Peak2Gene 分析的意义
Peak2Gene 分析通过整合单细胞 ATAC-seq 和 RNA-seq 数据,旨在揭示染色质可及性(Peak)与基因表达(Gene)之间的真实调控联系,是解析多组学调控网络的核心技术之一。
1. 基因调控网络简介
基因调控网络(Gene Regulatory Network, GRN)由转录因子(TF)、调控元件(如启动子、增强子、peaks)以及靶基因构成。TF 通过识别特定 DNA motif 结合到开放染色质区域,进而影响下游基因的转录。单细胞多组学手段的优势在于,可以在每个细胞分辨率下同时观测 TF 和靶基因的表达(scRNA-seq)、以及 TF 结合位点的染色质可及性(scATAC-seq,并结合 motif 识别进行推断)。
通过联合单细胞 ATAC 与 RNA 数据,可实现:
- TF 表达谱的确定
- TF 结合位点(peak)的开放性评估
- motif 的富集情况及功能活性推断
- 靶基因表达的动态变化监测
这种“TF 表达 × motif-peak 结合 × 靶基因表达”三维整合,有助于重建单细胞尺度的调控因果链,精准捕捉关键调控及作用靶点,并为 GRN 建图和机制深入提供着力点。
2. Peak2Gene分析如何助力解析GRN基因调控网络?
- 通过Motif 分析,我们可以知道TF 及 Peak 的调控关系。Motif 分析通过与已知转录因子识别序列比对,从开放染色质 peak 中识别可能被 TF 结合的区域,是精准描述 TF→peak 调控关系的重要基础。这些 motif 富集的 peaks 被视为 TF 调控潜在靶点,是调控网络的“上游”基础。具体Motif分析的相关内容请阅读Motif分析详解与实践流程
- Peak2Gene 分析则通过在单细胞水平考察 peak 可及信号与附近基因表达的相关性,精确捕捉每个 peak 与其潜在靶基因之间的调控配对,克服仅靠线性距离注释的局限。这一策略有力刻画了开放 peaks 实际参与基因调控的真实证据,阐释调控网络“下游”环节。
- 结合 motif 分析(TF→peak)与 Peak2Gene 分析(peak→gene),能够系统性描绘 TF、调控元件 peak 及其作用靶基因三者间的直接调控轴,为多组学调控机制研究提供多重证据支撑。
标准分析流程
- 差异 peak 筛选:定位具有生物学意义的开放区域;
- Peak2Gene 注释:整合表达及距离,建立 peak–gene 关联;
- Motif 分析:鉴定及评估 motif 富集和活性,预测关键 TF;
- 多组学联合验证:分析 motif 活性、TF/靶基因表达和 peak 可及性之间的调控一致性。
二、Peak2Gene 分析的实现
在 Signac 包中,主要通过LinkPeaks()核心函数实现peak-gene相关性分析: 对于每个基因,通过计算该基因表达量与其邻近 peaks 的可及性信号之间的相关性,并校正 GC 含量、整体可及性、peak 长度等多种技术偏置,从而识别出可能调控该基因的 peak 集合。
对于每个目标基因:
- 筛选距离该基因(如±500kb)范围内的 peaks;
- 计算这些 peaks 可及性与基因表达的相关性;
- 使用广义线性模型校正 GC%/peak size/整体可及性等偏置,获得更为可靠的 peak-gene 关联。
这种方式可以精确定量每个 peak 对靶基因的调控支持证据,是重构多组学调控网络的关键环节。实际项目落地时,推荐对感兴趣的基因集先行评估,也可扩展至全基因组自动注释。
# 1. 首先利用 RegionStats() 注释每个 peak 区域的 GC 含量等特征,用于后续偏置校正
DefaultAssay(obj) <- "ATAC"
obj <- RegionStats(obj, genome = BSgenome.Hsapiens.UCSC.hg38)
# 2. LinkPeaks(): 基于广义线性模型建立 peaks 与基因间相关性,并作多重偏置校正,得到显著 peak-gene 关联
obj <- LinkPeaks(
object = obj,
peak.assay = "ATAC",
expression.assay = "SCT",
genes.use = c("LYZ", "MS4A1") # 仅计算指定基因;如需全基因组可省略本参数
)三、Peak2Gene 结果展示
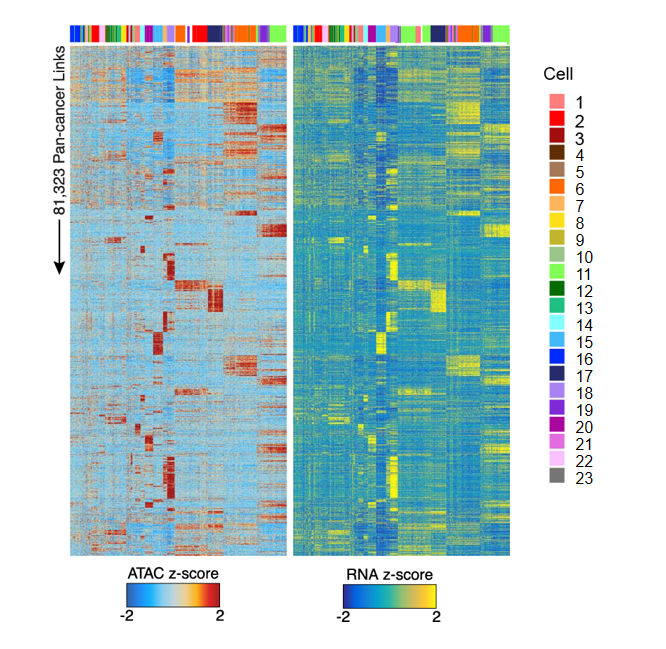
1. Peak-Gene 相关性热图
热图展示了染色质开放区域(peaks)与基因表达之间的相关性模式。热图分为两部分:
- ATAC热图(左):显示ATAC-seq信号强度(染色质可及性),蓝色表示低可及性,红色表示高可及性。
- RNA热图(右):显示相应基因的表达水平,蓝色表示低表达,黄色表示高表达。
- 每一行代表一个 peak-gene 对,每一列代表一个单细胞。
- 顶部颜色条标识不同细胞类型或群体,便于观察特定细胞群体中的peak-gene关联模式。
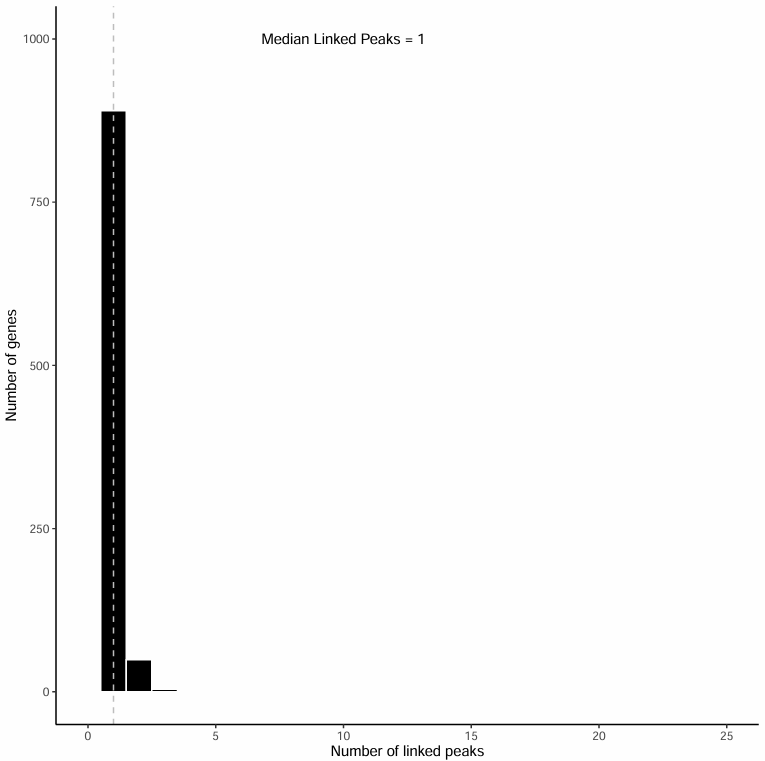
2. 关联peak数分布直方图
该直方图展示了每个基因关联到1~25个peak的数量分布:
- 横轴:基因所关联的peak数量(1~25)
- 纵轴:具有相应关联peak数的基因数量
- 虚线表示中位数位置,并以文本注明具体中位数
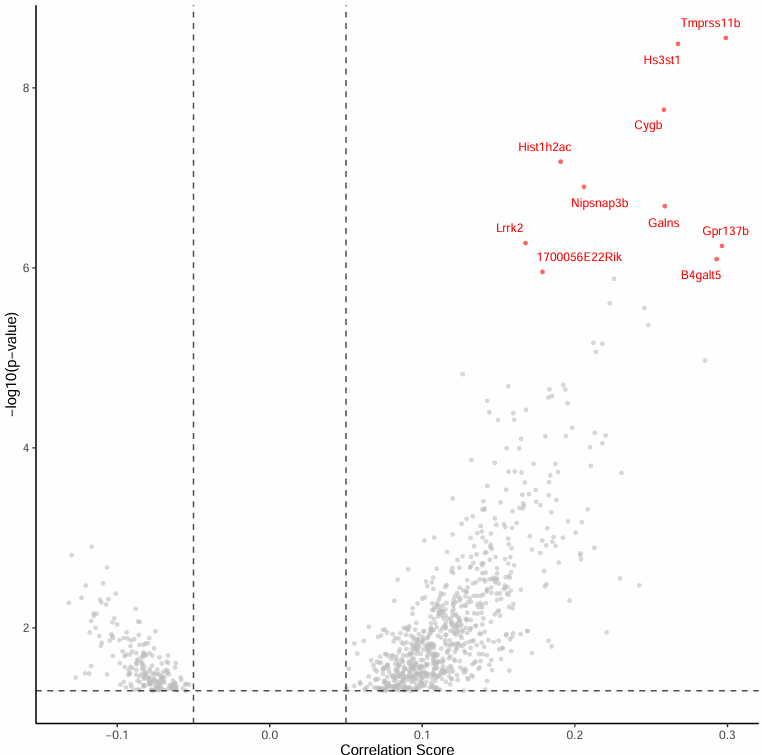
3. Top显著peak-gene关联火山图
火山图聚焦于Top 10最显著的peak-gene关联:
- 横轴:相关性score
- 纵轴:-log10(p)值
- 标注Top 10显著基因便于直观呈现
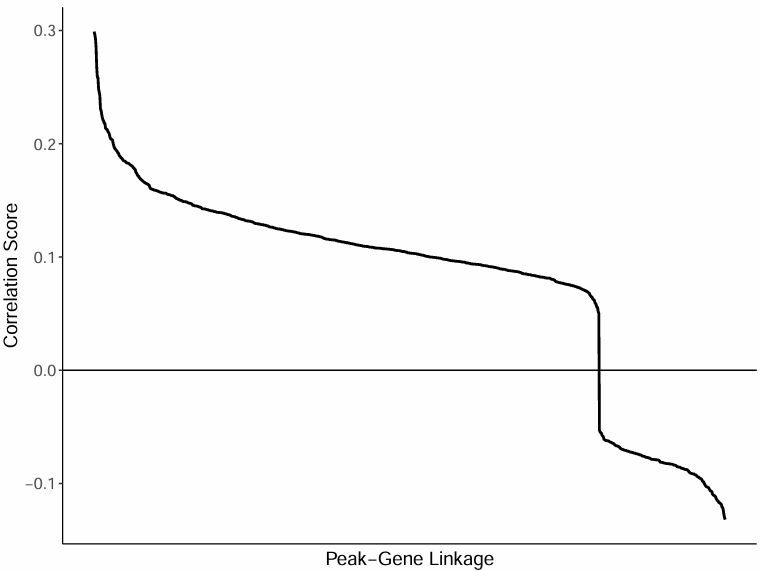
4. 全部peak-gene对相关性分布曲线
曲线图整体展示了全部peak-gene对的相关系数分布,可见相关性强的peak-gene对分布及趋势:
- 横轴:peak-gene对(按相关性高到低排序)
- 纵轴:对应相关系数
四. 常见问题
Q1:Peak2Gene 分析与 Motif 富集有什么联系?
A:两者关注角度不同,但结果可互补验证。
- Peak2Gene:主要回答“哪些 peak 实际调控了哪些基因”,通过相关性判定 peak-gene 调控关系,输出为 peak-gene 配对及相关性评分。
- Motif 富集:聚焦于“某一组 peak 中富集了哪些转录因子结合位点(motif)”,反映哪些 TF 可能调控这些 peak,输出为 motif 丰度及统计显著性。
Q2:为什么 Peak2Gene 得到的关联数较少?
A:可从以下方面排查原因:
- 样本或细胞数量较少,导致统计功效不足
- 选取的基因集过小或目标区域受限
- 设定的显著性阈值过高,过滤掉了较多关联
- RegionStats 步骤未正确执行,缺少偏置校正信息
- Motif 富集与 Peak2Gene 分析使用了不同细胞子集或过滤条件