血红蛋白基因表达污染处理建议
本指南总结了单细胞测序分析中遇到血红蛋白基因污染的常见处理思路和实用建议。
血红蛋白基因污染的来源与影响
在单细胞转录组测序(scRNA-seq)中,血红蛋白基因(如HBA1、HBA2、HBB等)高表达常见于外周血、骨髓等样本,主要来源于红细胞或其前体细胞。血红蛋白基因的高表达会导致下游分析(如聚类、差异表达分析)出现偏差,影响细胞类型注释的准确性[^1][^2]。
WARNING
血红蛋白基因污染可能导致非红细胞类群被错误聚类或注释,影响生物学结论的可靠性[^1]。
主流处理策略
分析前过滤
在数据预处理阶段,建议根据血红蛋白基因的表达比例(如每个细胞中血红蛋白基因转录本占比)过滤高污染细胞。例如,Seurat官方推荐去除血红蛋白基因表达比例高于一定阈值(如5%)的细胞[^1][^3]。去除血红蛋白基因再聚类
直接从表达矩阵中去除血红蛋白相关基因,重新进行高变基因筛选和聚类分析,有助于提升非红细胞类群的分辨率[^2][^4]。结合组织类型灵活决策
对于脾脏、骨髓等组织,红细胞前体细胞的存在具有生物学意义,不建议一刀切去除,应结合研究目的和组织特性灵活处理[^2][^5]。
NOTE
具体阈值和处理策略可根据样本类型、研究目的和下游分析需求调整[^1][^3][^5]。
案例一:去除血红蛋白基因后的分析影响
血红蛋白基因存在一定污染,但红细胞表达明显,细胞类型注释依然可以很好区分。
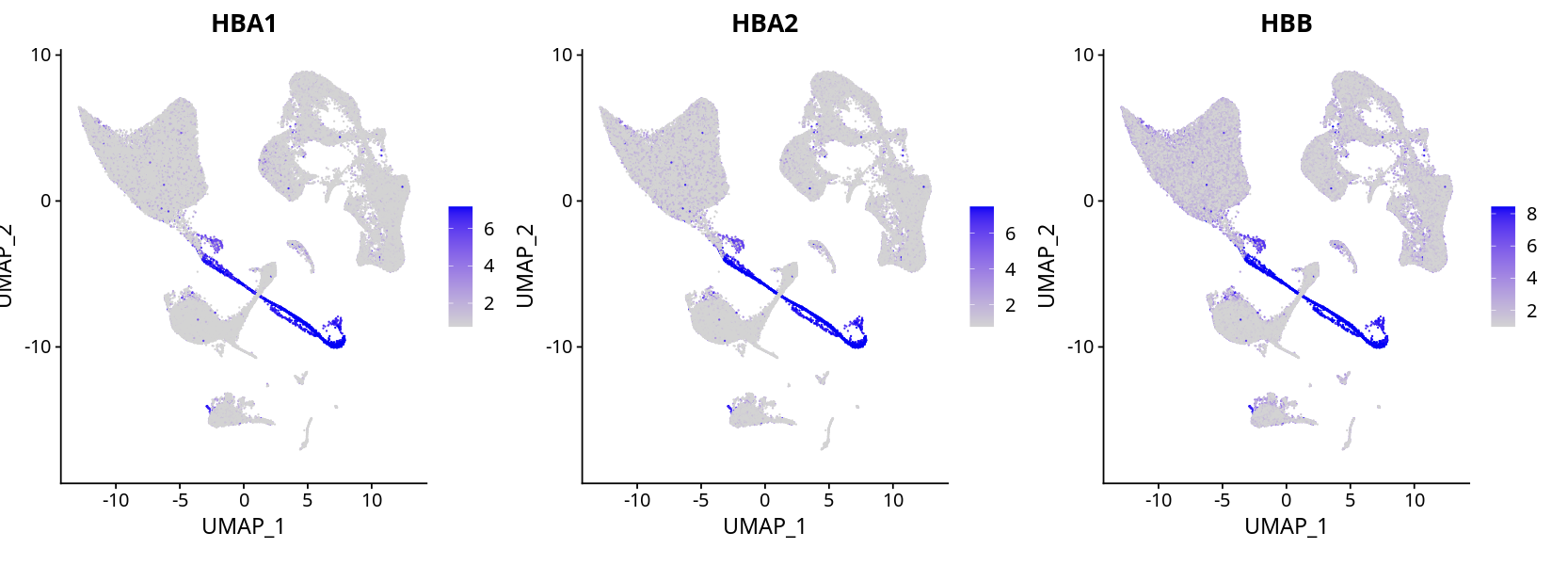
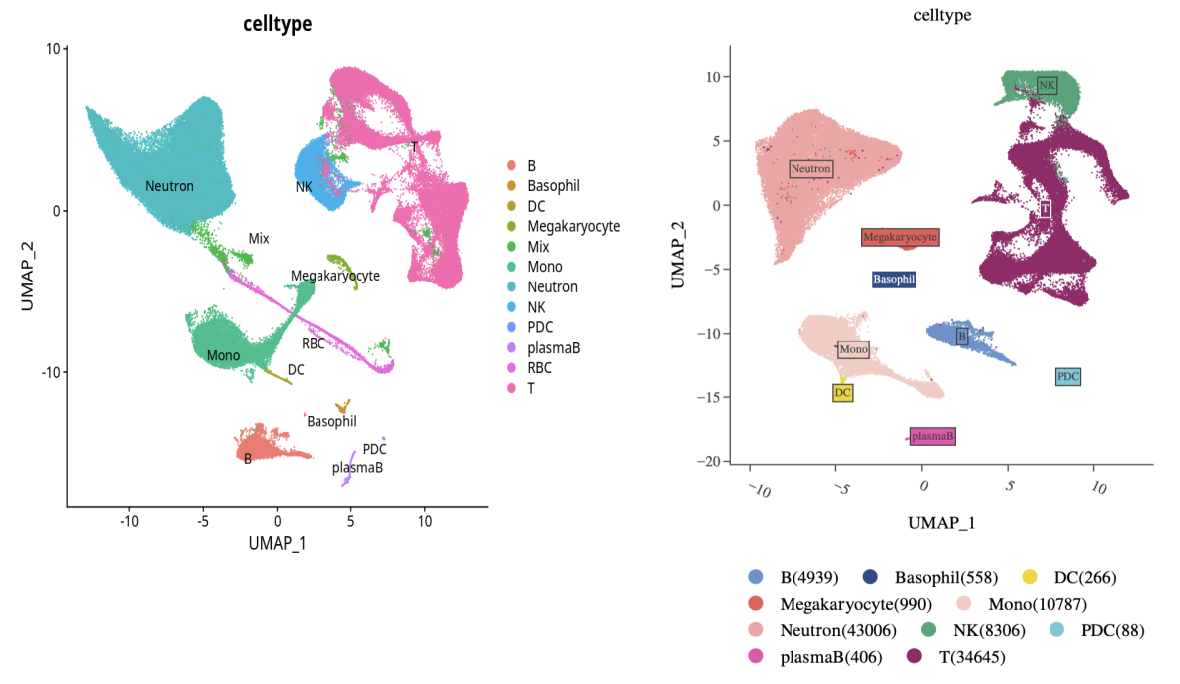
手动去除混合细胞群和红细胞,同时将矩阵中的血红蛋白基因去除,重新聚类后,细胞类型区分依然没有问题。
左:原聚类结果,右:去除血红蛋白基因后重新聚类
IMPORTANT
如不关注红细胞,可直接丢弃血红蛋白基因,重新聚类和高变基因查找。
TIP
整合分析时,建议先去除红细胞及其相关基因,再进行下游分析。
参考R代码:
counts <- GetAssayData(ob, assay = "RNA")
counts <- counts[-(which(rownames(counts) %in% c("HBA1","HBA2","HBB","HBD","HBE1","HBG1","HBG2","HBM","HBQ1","HBZ"))),]
obj <- subset(ob, features = rownames(counts))
DefaultAssay(obj) <- "RNA"
obj <- FindVariableFeatures(obj, selection.method = "vst", nfeatures = 2000, verbose = FALSE)
obj <- ScaleData(obj, verbose = FALSE)
obj <- RunPCA(obj, npcs = 30, verbose = FALSE)
obj <- FindNeighbors(obj, dims = 1:30)
obj <- FindClusters(obj, resolution = 0.5)
obj <- RunUMAP(obj, reduction = "pca", dims = 1:30)
obj <- RunTSNE(obj, reduction = "pca", dims = 1:30, check_duplicates = FALSE)案例二:红细胞前体细胞的识别与处理
整合注释,查看各个细胞类型marker gene的表达情况:
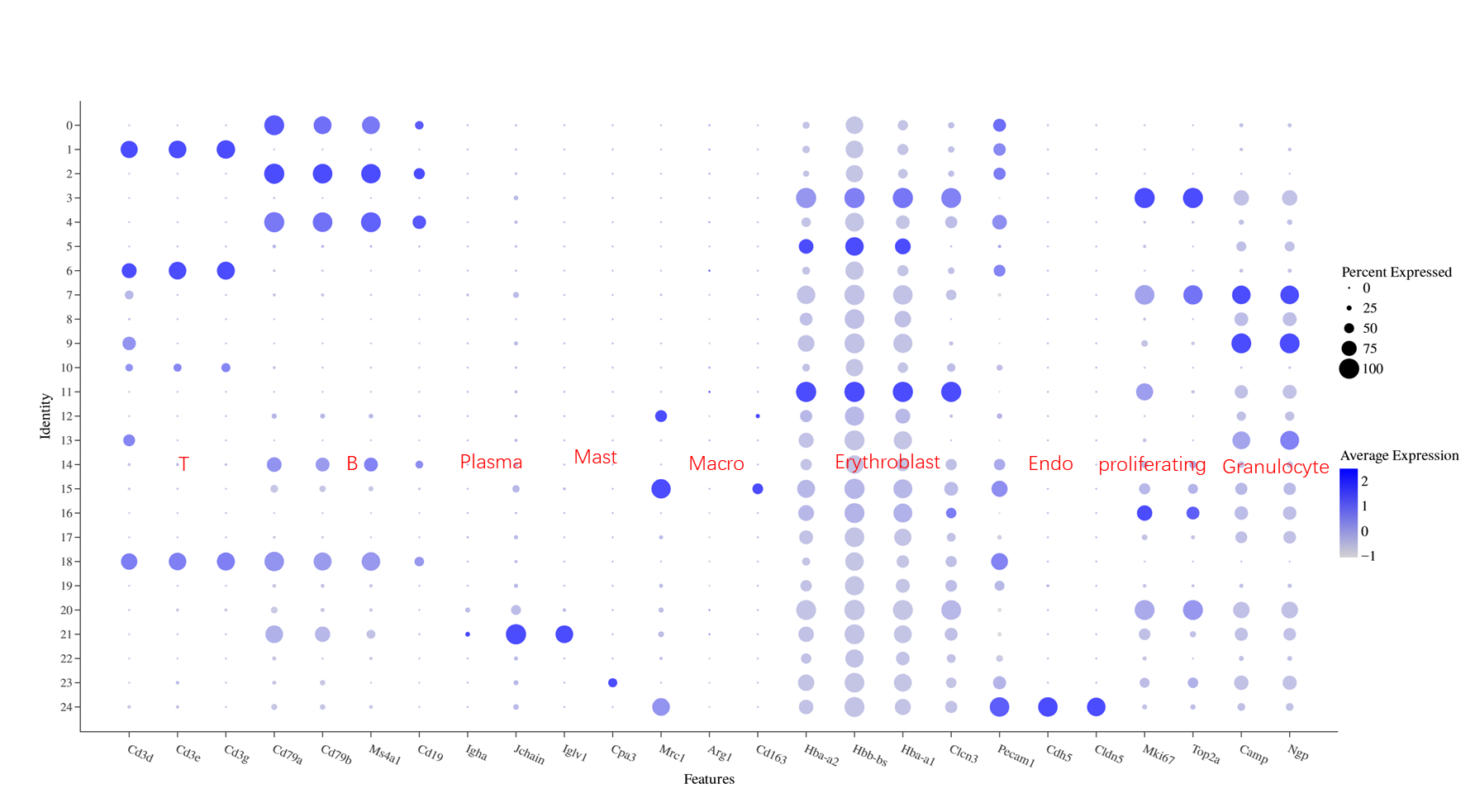
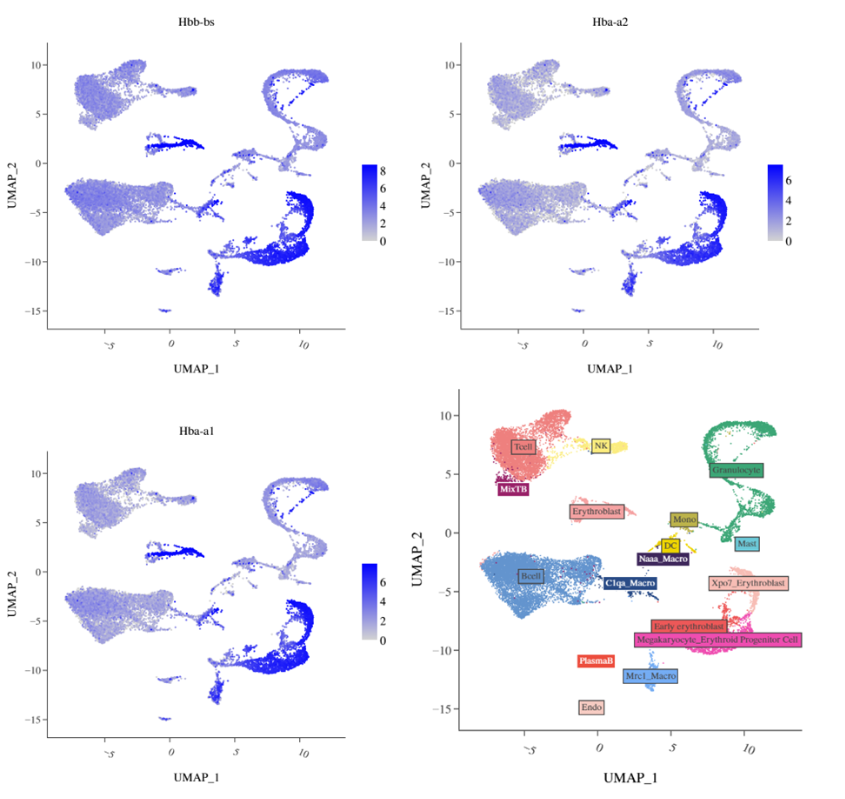
几乎每个cluster都表达血红蛋白marker基因,但部分cluster(umap右下)nFeature较高,可以特性注释为红细胞,其他细胞类型注释不受影响。进一步看这些细胞群表达增殖相关marker。
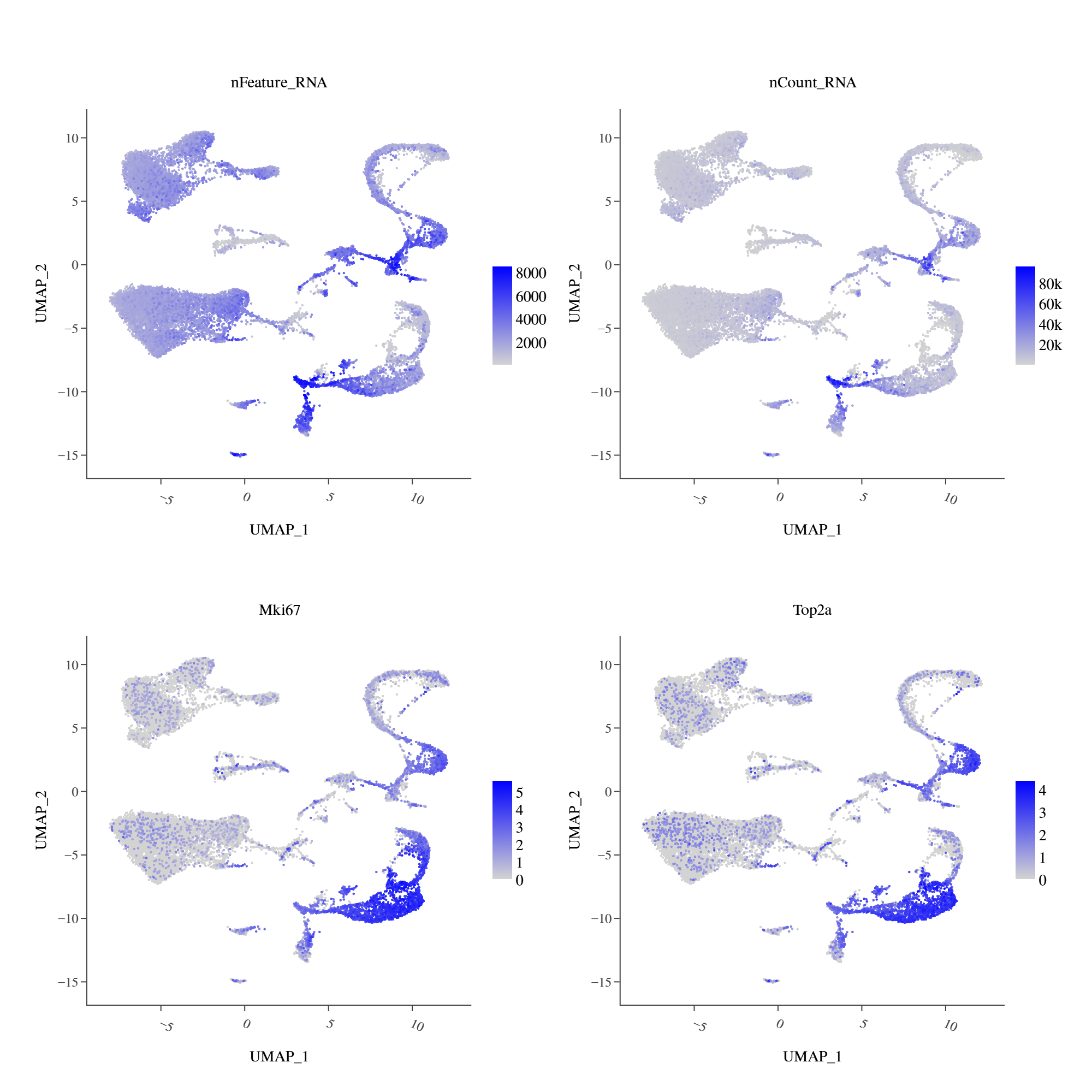
NOTE
这些红细胞可能为红细胞前体细胞。可以结合样本的具体组织特点进一步判断,比如脾脏、骨髓等组织,可能会存在红细胞发育相关的细胞类型,不建议一刀切直接丢掉。
TIP
红细胞前体细胞的去留建议结合组织类型和研究目的灵活决策。
总结与建议
IMPORTANT
- 不关注红细胞时,建议直接去除血红蛋白相关基因(如HBA1、HBA2、HBB等)及红细胞群体。
- 去除后重新聚类和高变基因查找,通常不会影响其他细胞类型的注释和分析。
- 某些特殊组织类型(如脾脏、骨髓等)可能存在大量红细胞或红细胞前体细胞,建议结合研究目标和样本特性,灵活决定后续处理方案。
TIP
处理流程可灵活调整,建议结合项目实际需求和下游分析目标。
参考文献
[^1]: Hao, Y., Hao, S., Andersen-Nissen, E., et al. (2021). Integrated analysis of multimodal single-cell data. Cell, 184(13), 3573-3587.e29. https://doi.org/10.1016/j.cell.2021.04.048
[^2]: Pijuan-Sala, B., Griffiths, J.A., Guibentif, C., et al. (2019). A single-cell molecular map of mouse gastrulation and early organogenesis. Nature, 566(7745), 490-495. https://doi.org/10.1038/s41586-019-0933-9
[^3]: Stuart, T., Butler, A., Hoffman, P., et al. (2019). Comprehensive Integration of Single-Cell Data. Cell, 177(7), 1888-1902.e21. https://doi.org/10.1016/j.cell.2019.05.031
[^4]: Luecken, M.D., Theis, F.J. (2019). Current best practices in single‐cell RNA‐seq analysis: a tutorial. Molecular Systems Biology, 15(6), e8746. https://doi.org/10.15252/msb.20188746
[^5]: Ziegenhain, C., Vieth, B., Parekh, S., et al. (2017). Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell, 65(4), 631-643.e4. https://doi.org/10.1016/j.molcel.2017.01.023