scRNA-seq + scATAC-seq 多模态数据整合分析教程
本教程面向初学者,使用 Seurat 与 Signac 演示如何整合两种单细胞数据:
- scRNA-seq:测基因表达(每个细胞哪些基因在“开”)
- scATAC-seq:测染色质可及性(每个细胞哪些 DNA 区域“开放”)
整合的核心目标是:将已注释好的 scRNA-seq 细胞标签“迁移/传递”(label transfer)到 scATAC-seq 上,从而给 ATAC 细胞自动注释类型。随后可做一次“共嵌入”(co-embedding)把两类细胞放到同一降维空间,便于同图展示(主要用于可视化)。
使用本教程前,建议准备:
- 已完成注释的 scRNA-seq 数据(例如已有
cell_type或聚类标签) - 与 scATAC-seq 相同物种、相同/兼容的基因组版本与染色体命名(如 hg38、chr1 格式)
- 基本的 R 运行环境与适度内存
我们将学习如何:
- 数据读取:分别读取 scRNA-seq 和 scATAC-seq 数据
- 数据预处理:进行质量控制(QC)与初步筛选
- 数据标准化:为后续整合打基础(RNA 的归一化与可变基因选择;ATAC 的 TF-IDF/LSI)
- 基因活性分数:将 ATAC 的 peaks 转为近似“表达”的基因活性矩阵
- 跨模态整合:用锚点匹配进行标签传递(label transfer)
- 可视化与导出:UMAP/共嵌入展示与结果保存
#加载必要的R包
suppressPackageStartupMessages({
library(Seurat)
library(Signac)
library(EnsDb.Hsapiens.v86)
library(BSgenome.Hsapiens.UCSC.hg38)
library(biovizBase)
#library(BSgenome.Mmusculus.UCSC.mm10)
#library(EnsDb.Mmusculus.v79)
library(dplyr)
library(ggplot2)
library(patchwork)
})
# 设置随机种子
set.seed(1234)
# 设置Seurat选项(注意:8000 * 1024^2 实际上是8GB)
options(future.globals.maxSize = 8000 * 1024^2) # 8GBmy36colors <-c( '#E5D2DD', '#53A85F', '#F1BB72', '#F3B1A0', '#D6E7A3', '#57C3F3', '#476D87',
'#E95C59', '#E59CC4', '#AB3282', '#23452F', '#BD956A', '#8C549C', '#585658',
'#9FA3A8', '#E0D4CA', '#5F3D69', '#C5DEBA', '#58A4C3', '#E4C755', '#F7F398',
'#AA9A59', '#E63863', '#E39A35', '#C1E6F3', '#6778AE', '#91D0BE', '#B53E2B',
'#712820', '#DCC1DD', '#CCE0F5', '#CCC9E6', '#625D9E', '#68A180', '#3A6963',
'#968175', "#6495ED", "#FFC1C1",'#f1ac9d','#f06966','#dee2d1','#6abe83','#39BAE8','#B9EDF8','#221a12',
'#b8d00a','#74828F','#96C0CE','#E95D22','#017890')1. 数据读取和准备
请准备以下文件:
scRNA-seq 数据(表达矩阵)
├── RNA_sample
│ ├── filtered_feature_bc_matrix
│ │ ├── barcodes.tsv.gz(细胞条形码)
│ │ ├── features.tsv.gz(基因列表)
│ │ └── matrix.mtx.gz(稀疏表达矩阵)
scATAC-seq 数据(peaks 矩阵 + 片段)
├── ATAC_sample
│ ├── filtered_peaks_bc_matrix
│ │ ├── barcodes.tsv.gz(细胞条形码)
│ │ ├── features.tsv.gz(peaks 列表)
│ │ └── matrix.mtx.gz(稀疏计数矩阵)
│ ├── ATAC_sample_fragments.tsv.gz(每条测序读段的基因组坐标,供 TSS 富集/核小体信号等 QC 使用)
│ ├── ATAC_sample_fragments.tsv.gz.tbi(fragments 的索引文件)
│ └── per_barcode_metrics.csv(可选的每细胞质控指标)
小贴士:
- 若使用不同平台/流程生成的数据,请确认能读入为等价的稀疏矩阵与片段文件。
- 尽量保证 RNA 与 ATAC 来自相近的样本/条件,便于标签传递更可靠。
获取基因注释信息
我们将从 EnsDb 数据库获取人类基因组注释(基因位置、转录本、外显子、TSS 等)。这些信息用于:
- 计算 ATAC 的 TSS 富集(判断开放染色质是否在转录起始位点附近更集中)
- 构建“基因活性矩阵”(把 peaks 信号映射到基因上)
请确保:
- 物种与参考基因组版本匹配(如
EnsDb.Hsapiens.v86对应 hg38) - 染色体命名风格一致(例如都用
chr1、chr2这样的前缀)
# 获取基因注释信息(静默处理警告和消息)
suppressWarnings({
suppressMessages({
annotation <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
seqlevels(annotation) <- paste0('chr', seqlevels(annotation))
genome(annotation) <- 'hg38'
})
})
# 设置并行计算(静默处理,可选)
suppressPackageStartupMessages({
library(future)
})
plan("multicore", workers = 4)读取 scRNA-seq 数据
我们将读入单细胞转录组的表达矩阵(barcodes/features/matrix 三件套),并创建标准的 Seurat 对象:
counts:每个细胞的基因计数meta.data:后续会加入样本信息、质控指标、聚类/细胞类型等
完成后得到的对象将作为“参考”(reference),用于给 ATAC 细胞传递标签。
options(warn = -1)
# 定义scRNA-seq数据路径
rna_sample_name <- 'joint34'
rna_counts_path <- file.path(rna_sample_name, 'filtered_feature_bc_matrix')
# 读取scRNA-seq数据
rna_counts <- Read10X(rna_counts_path)
# 创建scRNA-seq Seurat对象
rna_obj <- CreateSeuratObject(
counts = rna_counts,
project = "RNA_sample",
min.cells = 3,
min.features = 200
)
# 添加样本标识
rna_obj$orig.ident <- 'joint34'
rna_obj$Sample <- 'joint34'
rna_obj$technology <- 'RNA'
options(warn = 0)
# scRNA-seq数据读取完成读取 scATAC-seq 数据
这里会读入:
filtered_peaks_bc_matrix:peaks×cells 的稀疏计数矩阵(代表开放染色质计数)fragments.tsv.gz:每条读段的基因组坐标(用于 TSS 富集、核小体信号等 QC)
随后用 Signac 创建 ChromatinAssay 并包装为 Seurat 对象。这个对象将作为“查询”(query),接收来自 RNA 的标签传递。
# 定义scATAC-seq数据路径
atac_sample_name <- 'joint41'
atac_counts_path <- file.path(atac_sample_name, 'filtered_peaks_bc_matrix')
atac_fragments_path <- file.path(atac_sample_name, paste0(atac_sample_name, '_A_fragments.tsv.gz'))
#atac_metadata_path <- file.path(atac_sample_name, 'per_barcode_metrics.csv')
# 读取scATAC-seq数据
atac_counts <- Read10X(atac_counts_path)
# 读取质控指标metadata
#atac_metadata <- read.csv(
# file = atac_metadata_path,
# header = TRUE,
# row.names = 1
#)
# 创建ChromatinAssay对象
chrom_assay <- CreateChromatinAssay(
counts = atac_counts,
sep = c(':', '-'),
fragments = atac_fragments_path,
annotation = annotation,
min.cells = 3,
min.features = 200
)
# 创建scATAC-seq Seurat对象
atac_obj <- CreateSeuratObject(
counts = chrom_assay,
assay = "ATAC",
project = "ATAC_sample"#,meta.data = atac_metadata
)
# 添加样本标识
atac_obj$orig.ident <- 'joint41'
atac_obj$Sample <- 'joint41'
atac_obj$technology <- 'joint41'
# scATAC-seq数据读取完成2. 数据预处理
在整合前,需要先把两类数据分别“清洗干净”:
- QC(质量控制):剔除低质量细胞(如线粒体比例过高、ATAC 的 TSS 富集过低等)
- 初步标准化/降维:为后续找锚点、可视化做好准备
不同数据集的阈值会不同,下文会给出常见指标与可视化方法,帮助你按分布来调整阈值。
scRNA-seq 数据质量控制
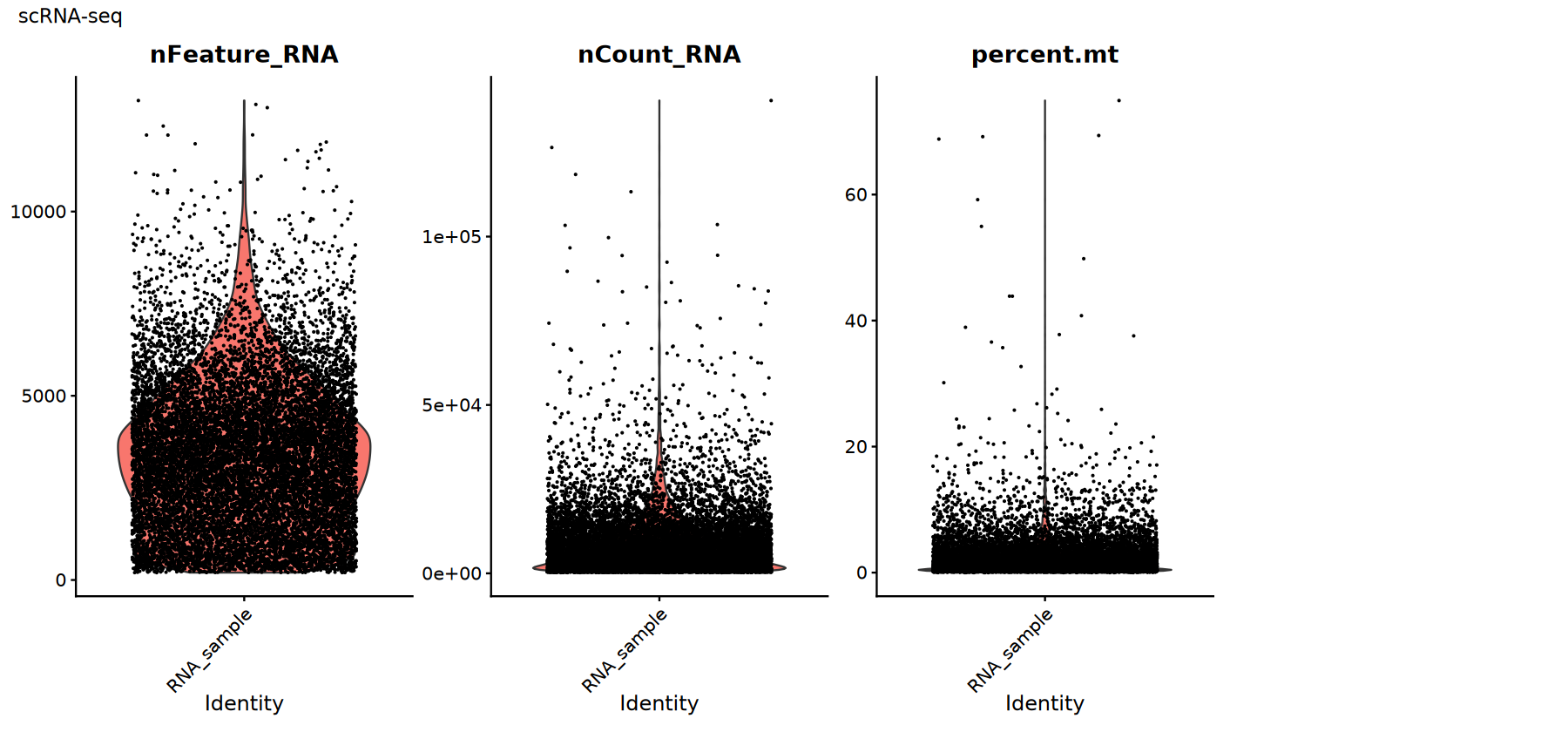
常用 QC 指标:
percent.mt:线粒体基因比例。过高通常提示细胞受损或 RNA 泄漏。nFeature_RNA:检测到的基因数。过低可能是空液滴,过高可能是双细胞/多重。nCount_RNA:总计数。与测序深度相关,需结合分布来判定异常。
这些指标没有“一刀切”的阈值,应结合数据分布(小提琴图/散点图)做经验性筛选。
# 计算线粒体基因表达比例
suppressWarnings({
rna_obj[["percent.mt"]] <- PercentageFeatureSet(rna_obj, pattern = "^MT-")
})scATAC-seq 数据质量控制
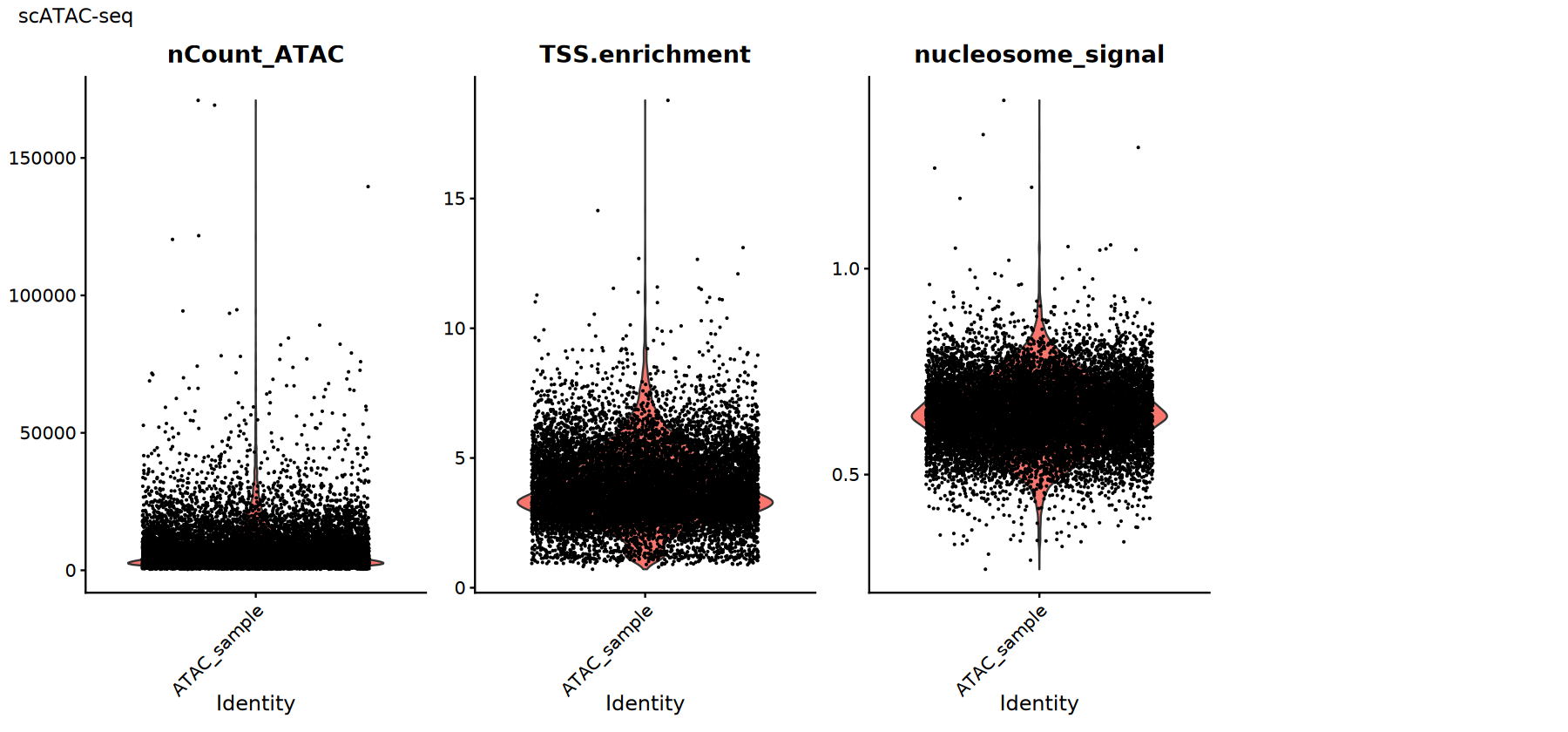
常用 QC 指标:
TSS.enrichment(TSS 富集分数):越高越好,说明信号在转录起始位点附近更集中;过低可能提示背景高或细胞活性差。nucleosome_signal(核小体信号):反映核小体周期性信号;通常越低越好。nCount_ATAC:总 ATAC 计数,极低或极高都需警惕异常。
实际阈值应基于数据分布设置,并与 fragments 质量、双细胞比例等因素结合判断。
# 设置默认assay为peaks
DefaultAssay(atac_obj) <- 'ATAC'
# 计算TSS富集分数
atac_obj <- TSSEnrichment(atac_obj)
# 计算核小体信号
atac_obj <- NucleosomeSignal(atac_obj)质量控制可视化
用小提琴图查看各 QC 指标的分布/异常点,帮助选择阈值:
- RNA:
nFeature_RNA、nCount_RNA、percent.mt - ATAC:
nCount_ATAC、TSS.enrichment、nucleosome_signal
建议:
- 观察是否存在明显的长尾或双峰分布
- 尝试多组阈值并比较下游聚类/UMAP 是否更清晰
# scRNA-seq质控指标可视化
suppressWarnings({
options(repr.plot.width = 15, repr.plot.height = 7)
p1 <- VlnPlot(rna_obj,
features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 4, pt.size = 0.1) +
plot_annotation(title = "scRNA-seq")
print(p1)
# scATAC-seq质控指标可视化
p2 <- VlnPlot(atac_obj,
features = c("nCount_ATAC", "TSS.enrichment", "nucleosome_signal"),
ncol = 4, pt.size = 0.1) +
plot_annotation(title = "scATAC-seq")
print(p2)
})
质量控制过滤
根据上一步的分布选择阈值并过滤低质量细胞。提示:
- 过滤后细胞数减少是正常现象,关键在于提升信噪比
- 请记录所用阈值与理由,便于复现与团队沟通
# scRNA-seq数据过滤
cat('过滤前scRNA-seq细胞数:', ncol(rna_obj), '\n')
rna_obj <- subset(rna_obj,
subset = nFeature_RNA > 200 &
nFeature_RNA < 10000 &
percent.mt < 20)
cat('过滤后scRNA-seq细胞数:', ncol(rna_obj), '\n')
# scATAC-seq数据过滤
cat('过滤前scATAC-seq细胞数:', ncol(atac_obj), '\n')
atac_obj <- subset(atac_obj,
subset = nCount_ATAC > 500 &
nCount_ATAC < 50000 &
TSS.enrichment > 1 &
nucleosome_signal < 1)
cat('过滤后scATAC-seq细胞数:', ncol(atac_obj), '\n')过滤后scRNA-seq细胞数: 13013
过滤前scATAC-seq细胞数: 13109
过滤后scATAC-seq细胞数: 12931
数据标准化处理
我们将分别对两类数据做适配的标准化/降维:
- RNA:Normalize → 选择可变基因 → Scale → PCA/UMAP → 邻居图与聚类
- ATAC:TF-IDF → 选重要特征(peaks)→ LSI(类似 PCA 的方法)→ UMAP
目的:把不同测序深度、计数尺度的样本拉到可比较的空间里,并初步揭示主轴结构,方便后续找“跨模态锚点”。
# scRNA-seq数据标准化处理
suppressWarnings({
suppressMessages({
rna_obj <- NormalizeData(rna_obj)
rna_obj <- FindVariableFeatures(rna_obj,nfeatures=4000)
rna_obj <- ScaleData(rna_obj)
rna_obj <- RunPCA(rna_obj)
rna_obj <- RunUMAP(rna_obj, dims = 1:30)
rna_obj <- FindNeighbors(rna_obj, dims = 1:30)
rna_obj <- FindClusters(rna_obj, resolution = 0.5)
})
})#scATAC-seq数据标准化处理
suppressWarnings({
suppressMessages({
atac_obj <- RunTFIDF(atac_obj)
atac_obj <- FindTopFeatures(atac_obj, min.cutoff = "q0")
atac_obj <- RunSVD(atac_obj)
atac_obj <- RunUMAP(atac_obj, reduction = "lsi", dims = 2:30, reduction.name = "umap.atac", reduction.key = "atacUMAP_")
})
})3. 基因活性分数计算
为了把 ATAC 的“开放程度”与 RNA 的“表达水平”联通起来,我们需要把 peaks 信号映射到基因上,得到“基因活性分数”(gene activity)。它不是直接的表达量,但在统计意义上可以近似反映某个基因的潜在表达活性,是跨模态整合的关键桥梁。
计算基因活性矩阵
思路:统计与每个基因相关的 peaks 信号(如基因体 + 上下游一定范围的启动子区域),累积得到该基因在每个细胞的“活性分数”。
- 典型做法会取 TSS 上游 ~2kb、下游 ~1kb 的窗口(可按需要调整)
- 得到的
ACTIVITYassay 将用于与 RNA 的RNAassay 建立锚点
# 计算基因活性分数
# 使用基因体和启动子区域计算活性分数
suppressWarnings({
DefaultAssay(atac_obj)="ATAC"
gene.activities <- GeneActivity(
object = atac_obj,
features = VariableFeatures(rna_obj),extend.upstream = 2000,extend.downstream = 1000)
# 将基因活性矩阵添加为新的assay
atac_obj[['ACTIVITY']] <- CreateAssayObject(counts = gene.activities)
DefaultAssay(atac_obj) <- "ACTIVITY"
# 标准化基因活性数据
atac_obj <- NormalizeData(atac_obj)
atac_obj <- ScaleData(atac_obj, features = rownames(atac_obj))
})4. 多模态数据整合
现在我们用“RNA 表达矩阵(参考)”与“ATAC 基因活性矩阵(查询)”进行跨模态匹配。核心任务:
- 找到跨模态的“对应关系”(锚点)
- 把“参考”的细胞标签传递给“查询”细胞
寻找整合锚点
“锚点”(anchors)是把两个数据空间对齐的关键。做法是用 CCA/LSI 等方法在 RNA 与 ATAC(基因活性)之间寻找一组高可比性的特征子空间,从而建立可互相投影的坐标系。
# 设置scATAC-seq对象的默认assay为ACTIVITY(基因活性分数)
DefaultAssay(atac_obj) <- 'ACTIVITY'
suppressWarnings({
suppressMessages({
# 寻找整合锚点
transfer.anchors <- FindTransferAnchors(
reference = rna_obj,
query = atac_obj,
features = VariableFeatures(object = rna_obj),
reference.assay = 'RNA',
query.assay = 'ACTIVITY',
k.anchor = 30,
k.filter = 50 ,
reduction = 'cca'
)
})
})预测细胞类型标签
当锚点建立后,就可以把“参考”的标签(如 cell_type 或 seurat_clusters)传给 ATAC 细胞:
- 输出的
predicted.id即为每个 ATAC 细胞的预测类型/聚类 - 可同时得到预测得分,用于衡量置信度(本教程示例显示聚类标签)
建议:
- 用 UMAP 查看预测分布是否与 RNA 的结构一致
- 结合标记基因/已知生物学知识做 sanity check
# 预测scATAC-seq细胞的聚类标签
predicted.labels <- TransferData(
anchorset = transfer.anchors,
refdata = rna_obj$seurat_clusters,#scRNA-seq数据实际细胞注释结果,按照scRNA-seq实际注释情况选择
weight.reduction = atac_obj[['lsi']],
dims = 2:30
)
# 将预测标签添加到scATAC-seq对象
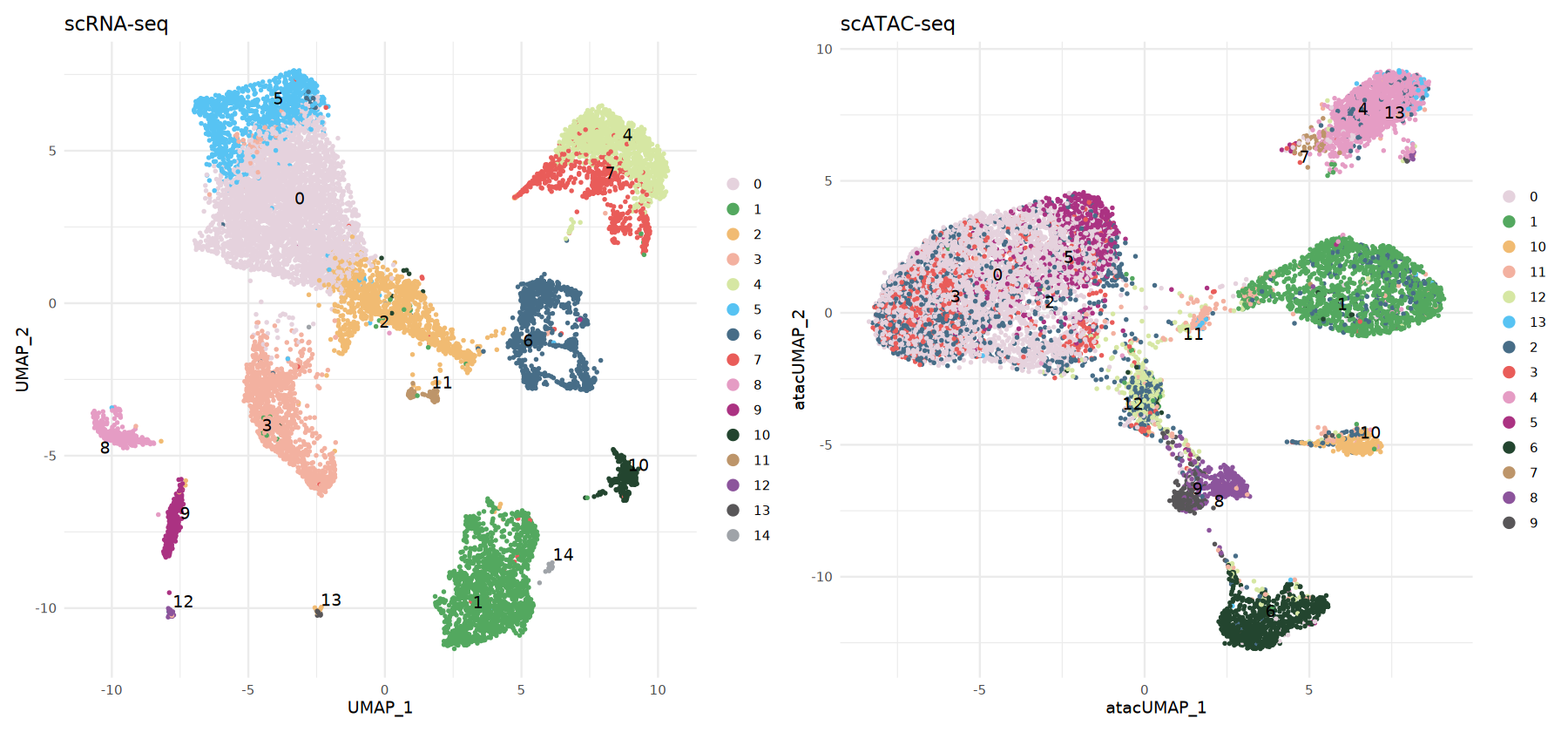
atac_obj <- AddMetaData(atac_obj, metadata = predicted.labels)可视化 scATAC-seq 细胞类型
这里并排展示:
- RNA:参考数据自带/计算得到的聚类
- ATAC:通过标签传递得到的
predicted.id
颜色对照相同类别,便于直接比较两者结构的一致性。
# scRNA-seq数据可视化
options(repr.plot.width = 15, repr.plot.height = 7)
p1 <- DimPlot(rna_obj,
group.by = 'seurat_clusters',
label = TRUE,
repel = TRUE,
pt.size = 0.5) +
ggtitle('scRNA-seq') +
theme_minimal() +
scale_color_manual(values = my36colors)
# scATAC-seq数据可视化(基于预测标签)
p2 <- DimPlot(atac_obj,
group.by = 'predicted.id',
label = TRUE,
repel = TRUE,
pt.size = 0.5) +
ggtitle('scATAC-seq') +
theme_minimal() +
scale_color_manual(values = my36colors)
p1 | p2
pdf("scRNA_scATAC_UMAP.pdf", width = 10, height = 5)
print(p1 | p2)
dev.off()pdf: 2
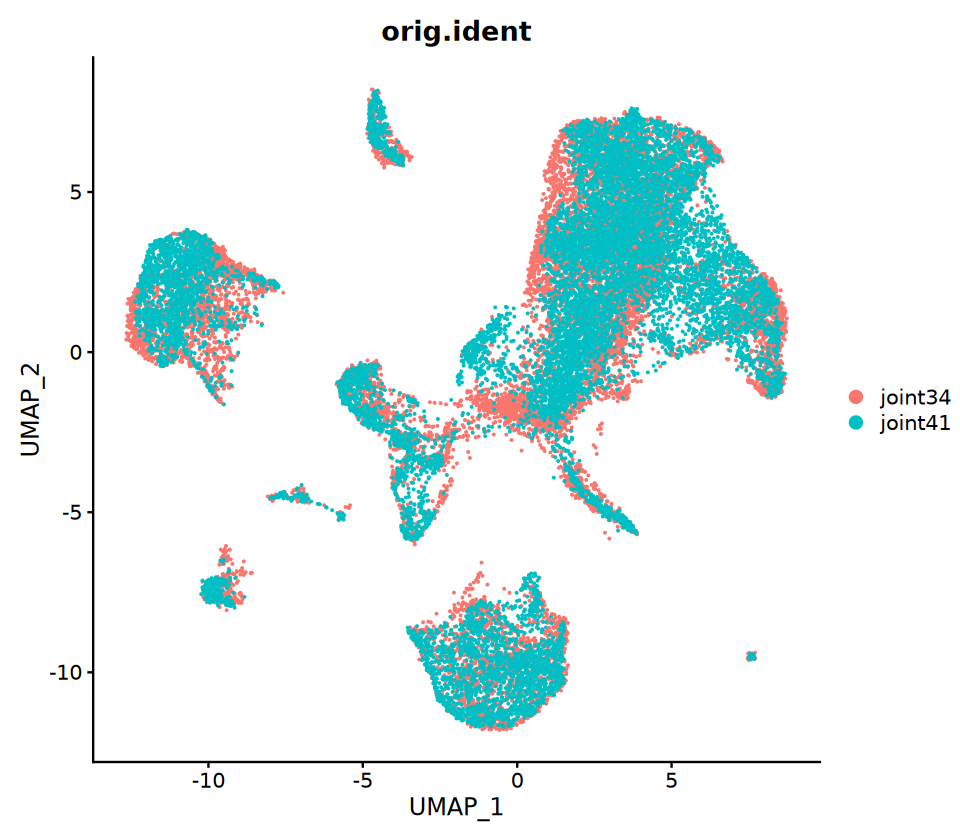
5.共嵌入分析(可选)
目的:把 RNA 与 ATAC 细胞放到同一张 UMAP 上,便于观察跨模态的一致性。 方法要点:
- 先用锚点把 RNA 的表达矩阵“插补/投影”到 ATAC 上,得到 ATAC 细胞的“拟表达矩阵”
- 把两者合并后再做统一的降维与 UMAP
注意:共嵌入主要是可视化工具,不等同于真实的同细胞多组学观测,解读时请结合生物学常识。
# 计算共嵌入
suppressWarnings({
suppressMessages({
genes.use <- VariableFeatures(rna_obj)
refdata <- GetAssayData(rna_obj, assay = "RNA", slot = "data")[genes.use, ]
# refdata (input) contains a scRNA-seq expression matrix for the scRNA-seq cells. imputation
# (output) will contain an imputed scRNA-seq matrix for each of the ATAC cells
imputation <- TransferData(anchorset = transfer.anchors, refdata = refdata, weight.reduction = "cca",dims=1:30)
atac_obj[["RNA"]] <- imputation
coembed <- merge(x = rna_obj, y = atac_obj)
coembed <- ScaleData(coembed, features = genes.use, do.scale = FALSE)
coembed <- RunPCA(coembed, features = genes.use, verbose = FALSE)
coembed <- RunUMAP(coembed, dims = 1:30)
})
})
options(repr.plot.width = 8, repr.plot.height = 7)
p3=DimPlot(coembed, group.by = c("orig.ident"))
p3
pdf("coembed_UMAP.pdf", width = 10, height = 5)
print(p3)
dev.off()pdf: 2
6. 结果总结和保存
完成后请保存关键对象,便于复现与后续分析:
scRNA_processed.rds:已标准化/降维/聚类的 RNA 对象scATAC_processed.rds:已 QC/标准化/带预测标签的 ATAC 对象(含ACTIVITY)coembed.rds:共嵌入对象(若执行了该步骤)session_info.txt:运行环境信息,方便他人复现
# 保存分析对象
saveRDS(rna_obj, file = "./scRNA_processed.rds")
saveRDS(atac_obj, file = "./scATAC_processed.rds")
saveRDS(coembed, file = "./coembed.rds")7. 进阶分析建议
在完成基础整合后,你可以进一步深入:
7.1 转录因子 motif 分析
- 用
FindMotifs()在差异可及性区域(DAR)中寻找富集的 TF motif - 结合 RNA 的表达或基因活性推断 TF 活性变化
7.2 基因调控网络(GRN)与 peaks-to-gene 连接
- 用
LinkPeaks()识别 peaks 与靶基因的潜在调控关系 - 若仅有单组学数据,可考虑 imputation 或 pseudo-bulk 建立表达与可及性的对应;最佳做法是一胞多组学数据进行直接验证
7.3 发育轨迹/伪时分析
- 在整合空间上进行细胞轨迹推断
- 对比基因表达与可及性沿轨迹的动态变化
7.4 差异可及性与差异表达
- 比较不同细胞类型/状态的差异 peaks 与差异基因
- 识别细胞类型特异的调控元件
7.5 功能富集
- 对差异基因或差异 peaks 关联基因做 GO/KEGG 等富集
- 结合已知通路解释细胞功能特征