全序列可变剪接分析:rMATS 流程与示例脚本
介绍
NOTE
可变剪接(alternative splicing)又称作选择性剪接,是指在真核生物的基因转录过程中,前体RNA经过不同的剪接形式,产生多个不同的转录本。在这个剪接过程中,不同的外显子和内含子可以选择性地被剪除或保留。
IMPORTANT
寻因研发的全序列测序技术(scFAST-seq),是基于随机引物来捕获RNA分子,在单细胞的转录组水平上实现了全长覆盖,从而为可变剪接分析创造了机会和可能。这项转录组测序技术是基于droplet-based原理的高通量测序技术,因此,我们更建议在细胞群水平或样本水平上进行可变剪接分析。
rMATS 分析软件
软件介绍
我们使用了 rMATS 来分析可变剪接,rMATS(Replicate Multivariate Analysis of Transcript Splicing)是一个基于 RNA-seq 数据来分析差异性可变剪接事件的软件。rMATS 可以识别不同类型的可变剪接事件,包含 SE,A5SS,A3SS,MXE 和 RI 五种类型。
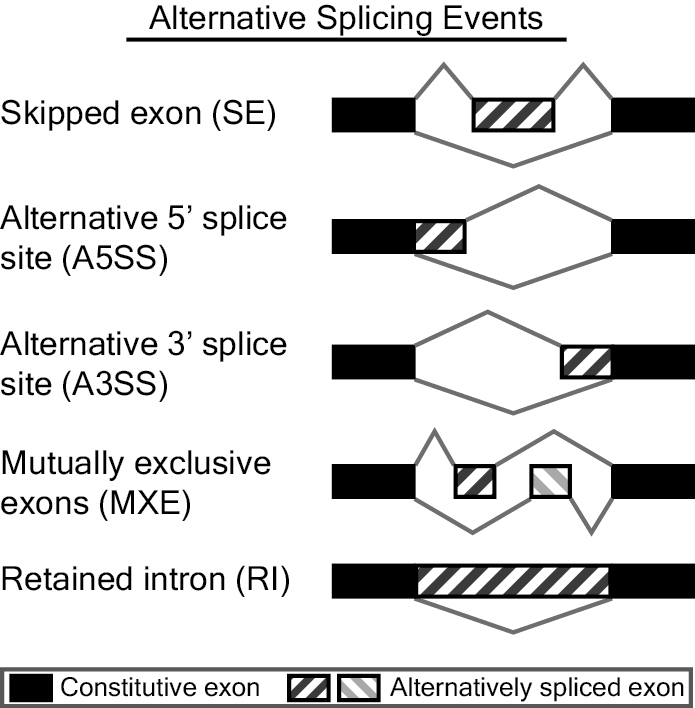
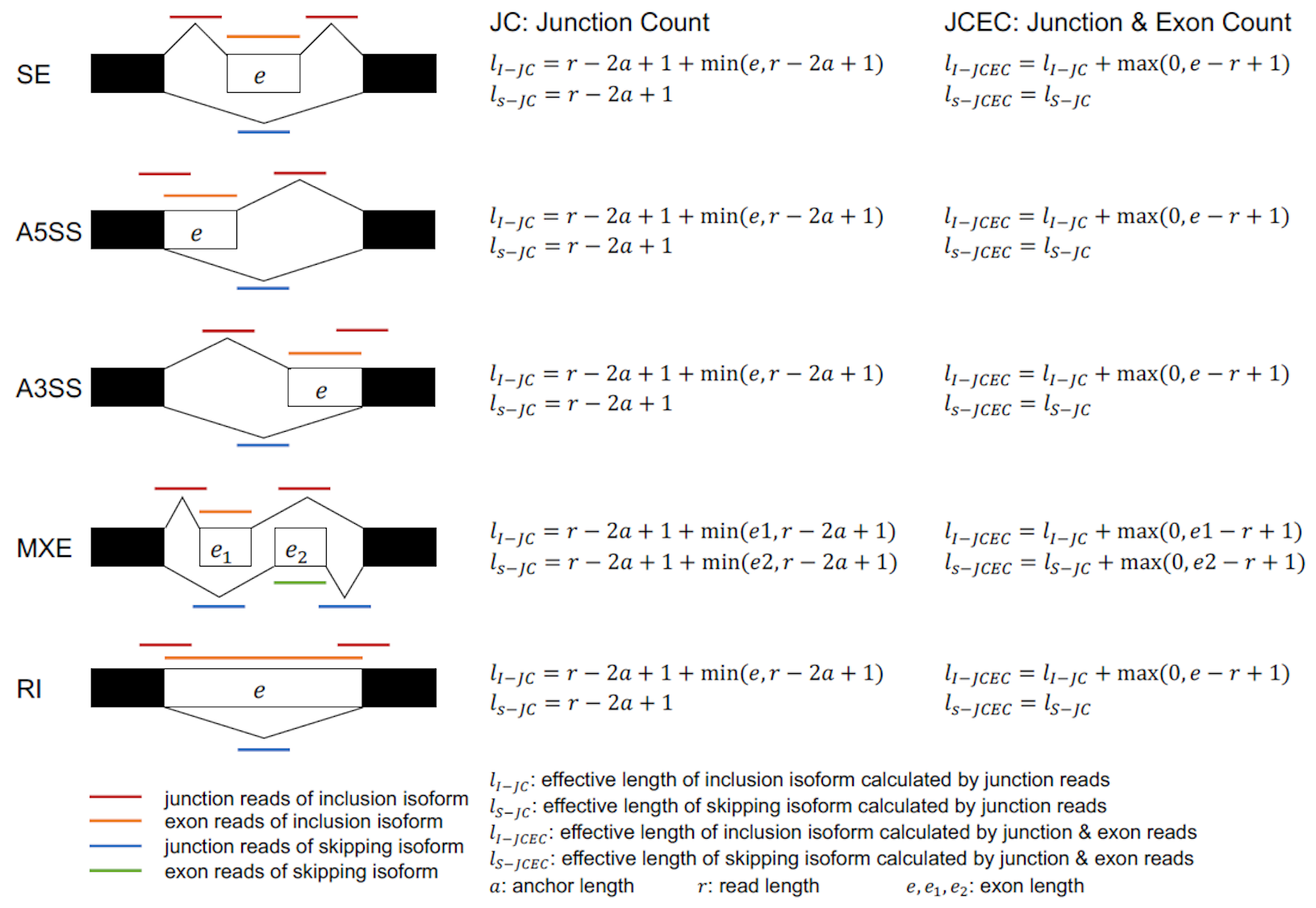
对识别到的可变剪接事件,rMATS 分别按照两种不同的计算方式来统计支持 Reads 数。(JC,Junction Counts;JCEC,Junction Counts and Exon Counts)
软件安装
Conda 安装 rMATS
TIP
推荐使用 Conda 来安装 rMATS 软件,首先创建一个新的 Conda 环境,您也可以选择激活已有环境:
conda create -n rMATS_env
conda activate rMATS_env安装:
conda install bioconda::rmats运行成功后,rMATS 就安装好了。
配置环境
IMPORTANT
运行后续代码,需要配置一些必要的工具,按照以下示例分别安装 Python 包和 R 包:
# install python packages
python -m pip install pysam click
# install R
conda install r-base
# install R packages
Rscript -e 'install.packages(c("argparse", "Seurat"))''至此,分析环境已经配置完成了!
示例数据和脚本下载
在这里为您提供了相应的示例数据和分析脚本,来帮助您理解后续的分析流程。该数据均来源于寻因官网 PBMC 数据。
wget https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/rMATS_demo.tar.gz
tar -zxvf rMATS_demo.tar.gzNOTE
示例数据包含 3 个文件:
PBMC.rds是基于 SingleCellExperiment 格式的单细胞数据PBMC_chr1.bam是从 PBMC 样本测序数据与 GRCh38 基因组的比对结果中抽取了 1 号染色体上的比对数据作为示例genes.gtf是基于 GRCh38 版本的基因组注释文件
三个脚本中,getbarcode.R 是用于读取 RDS 的细胞 barcode 信息和分组信息;rmats_run.py 首先根据指定的分组信息对 BAM 文件进行拆分,然后使用 rMATS 来识别可变剪接事件,并在指定分组之间计算差异表达水平。run.sh 是直接调用的运行脚本。
运行示例
CAUTION
运行以下命令前,请确保已激活环境:
conda activate rMATS_env运行以下命令,完成可变剪接分析:
# create output path
mkdir -p /path/result
# run
python /path/rmats_run.py \
--samplename demo \
--rds /path/demodata/PBMC.rds \
--outdir /path/result \
--bam /path/demodata/PBMC_chr1.bam \
--gtf /path/demodata/genes.gtf \
--cellanno seurat_clusters上述参数含义为:
| 参数名称 | 参数含义 |
|---|---|
| samplename | 样本名称 |
| rds | 基于 SingleCellExperiment 格式的数据,Seurat 对象 |
| outdir | 输出目录 |
| bam | 比对结果文件 |
| gtf | 基因组注释文件 |
| cellanno | 指定的 metadata 名,需存在于 RDS 里。注:cellanno 列不能包含 "/" |
TIP
小编用示例数据运行,在 4 core 16 GB 的环境下,运行了 28 分钟左右。
结果解读
运行完成后得到了哪些结果?又该如何理解呢?接下来,为您简单介绍结果内容。
首先,查看结果目录,可以看到输出结果按照指定的 cluster 分为了不同的文件夹,以 cluster0 为例:
tree -L 1 result/cluster0/
result/cluster0/
├── A3SS.MATS.JCEC.txt
├── A5SS.MATS.JCEC.txt
├── demo.b1.txt
├── demo.b2.txt
├── demo_cluster0_A3SS.MATS.JC.txt
├── demo_cluster0_A5SS.MATS.JC.txt
├── demo_cluster0_MXE.MATS.JC.txt
├── demo_cluster0_RI.MATS.JC.txt
├── demo_cluster0_SE.MATS.JC.txt
├── fromGTF.A3SS.txt
├── fromGTF.A5SS.txt
├── fromGTF.MXE.txt
├── fromGTF.novelJunction.A3SS.txt
├── fromGTF.novelJunction.A5SS.txt
├── fromGTF.novelJunction.MXE.txt
├── fromGTF.novelJunction.RI.txt
├── fromGTF.novelJunction.SE.txt
├── fromGTF.novelSpliceSite.A3SS.txt
├── fromGTF.novelSpliceSite.A5SS.txt
├── fromGTF.novelSpliceSite.MXE.txt
├── fromGTF.novelSpliceSite.RI.txt
├── fromGTF.novelSpliceSite.SE.txt
├── fromGTF.RI.txt
├── fromGTF.SE.txt
├── JCEC.raw.input.A3SS.txt
├── JCEC.raw.input.A5SS.txt
├── JCEC.raw.input.MXE.txt
├── JCEC.raw.input.RI.txt
├── JCEC.raw.input.SE.txt
├── JC.raw.input.A3SS.txt
├── JC.raw.input.A5SS.txt
├── JC.raw.input.MXE.txt
├── JC.raw.input.RI.txt
├── JC.raw.input.SE.txt
├── MXE.MATS.JCEC.txt
├── RI.MATS.JCEC.txt
├── SE.MATS.JCEC.txt
├── summary.txt
└── tmp
1 directory, 38 files上述 txt 文件是 rMATS 对 cluster0 相对其余 cluster 进行的差异可变剪切分析结果,各个文件介绍可以参考 rMATS 官方说明: [rmats-turbo/README.md at v4.3.0 · Xinglab/rmats-turbo (github.com)](https://github.com/Xinglab/rmats-turbo/blob/v4.3.0/README.md)
在此,为您介绍主要的输出结果内容:
主要结果 demo_cluster0_*.MATS.JC.txt
该结果分为 SE,A5SS,A3SS,MXE 和 RI 五个文件,分别对应 5 种可变剪接类型。
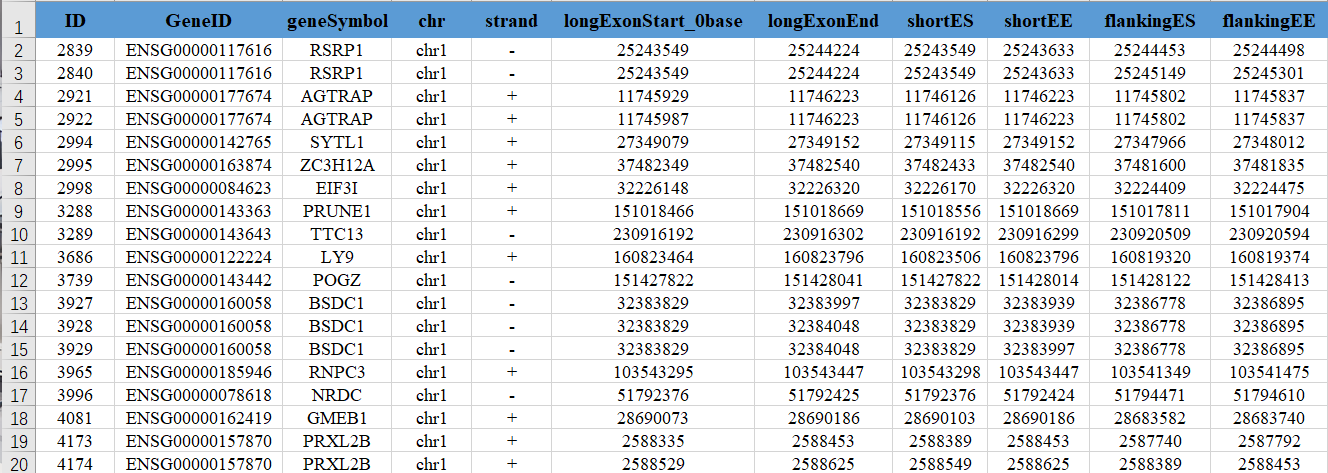
内容包含了各个剪接事件的起始结束、比对reads的详细信息,并就cluster0和其他所有cluster之间计算了各剪接事件的差异水平。各列描述为:
(1) ID:rMATS event id
(2) GenelD: 可变剪接事件所在基因编号
(3) geneSymbol: 可变剪接事件所在基因名称
(4) chr: 可变剪接事件所在染色体
(5) strand: 可变剪接事件所在染色体链的方向
(6) ExonStart_0base: 可变剪接事件跳跃外显子的起始位置,以0开始计数
(7) ExonEnd: 可变剪接事件跳跃外显子的终止位置
(8) upstreamES: 可变剪接事件跳跃外显子的上游exon起始位置
(9) upstreamEE: 可变剪接事件跳跃外显子的上游exon终止位置
(10) downstreamES: 可变剪接事件跳跃外显子的下游exon起始位置
(11) downstreamEE: 可变剪接事件跳跃外显子的下游exon终止位置
(12) ID:rMATS event id
(13) IJC_SAMPLE_1: 样本一在inclusion junction(IJC)下的count数,重复样本的结果以逗号分隔
(14) SJC_SAMPLE_1: 样本一在skipping junction(SJC)下的count数,重复样本的结果以逗号分隔
(15) IJC_SAMPLE_2: 样本二在inclusion junction(IJC)下的count数,重复样本的结果以逗号分隔
(16) SJC_SAMPLE_2: 样本二在skipping junction(SJC)下的count数,重复样本的结果以逗号分隔
(17) IncFormLen: 可变剪接事件 Exon Inclusion Isoform 的有效长度
(18) SkipFormLen: 可变剪接事件 Exon Skipping Isoform 的有效长度
(19) PValue: 两组样本间可变剪接事件表达差异显著性 p 值
(20) FDR: 可变剪接事件表达差异显著性 FDR 值
(21) IncLevel1: 处理组可变剪接事件 Exon Inclusion Isoform 在两个 Isoform 总表达量的比值
(22) IncLevel2: 对照组可变剪接事件 Exon Inclusion Isoform 在两个 Isoform 总表达量的比值
(23) IncLevelDifference: IncLevel1 与 IncLevel2 的差值
主要结果 summary.txt

该文件是对cluster0的差异可变剪接事件的汇总,统计了 FDR<0.05 的可变剪接事件数量。
其中 SignificantEventsJC/JCEC 是 SigEventsJC/JCECSample1HigherInclusion 和 SigEventsJC/JCECSample2HigherInclusion 的加和。JC和JCEC是指由两种不同计算方式各自得到的结果。
TIP
更多结果的查询和了解可以移步rMATS官网https://github.com/Xinglab/rmats-turbo/blob/v4.3.0/README.md
参考资料
[1] SHEN S, PARK J W, LU Z X, et al. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data[J]. PNAS, 2014, 111(51): E5593-601.