ATAC + RNA 多组学:Monocle3 拟时序与染色质动态解析
文档概览
Monocle3 拟时序分析是利用 Monocle3 工具基于 scATAC-seq 单组学数据来推断分化轨迹,拟时序分析用于重建细胞发育或分化过程的轨迹,揭示染色质可及性的动态变化。Monocle3 通过选择 root 细胞,计算每个细胞到起点的距离,得到伪时间(pseudotime),用于排序细胞的分化进程。在 scATAC-seq 分析中,伪时间可以直观反映染色质可及性的变化和基因调控网络的动态过程。
拟时序分析的意义
细胞分化与表观遗传动态
细胞分化是一个连续的过程,涉及表观遗传状态的渐进性改变。在单细胞分辨率下,我们可以观测到:
- 不同分化阶段的细胞同时存在于样本中
- 染色质可及性模式随分化进程发生系统性变化
- 关键调控元件(peaks)的开放/关闭状态与细胞命运决定密切相关
拟时序分析如何助力解析表观遗传动态
- 轨迹重建:通过降维和图形学习,Monocle3 能够识别细胞在表观遗传空间中的连续路径,将离散的细胞状态连接成有向的发育轨迹。
- 伪时间推断:基于指定的起始细胞(root cells),Monocle3 计算每个细胞沿轨迹到起点的最短路径距离,赋予每个细胞一个伪时间值,反映其在分化过程中的相对位置。
- 动态变化识别:通过比较不同伪时间点的染色质可及性模式,可以识别在分化过程中动态开放或关闭的调控元件,揭示关键转录因子结合位点的时序激活模式。
- 与多组学整合:拟时序分析结果可与 scRNA-seq 数据整合,揭示染色质可及性变化与基因表达变化的时序关联,构建动态的基因调控网络。
TIP
scRNA-seq 与 scATAC-seq:如何选择拟时序分析数据类型?
一胞双组学同时获得 scRNA-seq 和 scATAC-seq 两类数据,两者均可进行拟时序分析,不过它们反映的生物学层面有所区别:
- scRNA-seq:主要反映基因表达层面的动态变化,更适合用来刻画细胞功能和表型的转变—— 侧重“结果”视角。
- scATAC-seq:揭示染色质可及性的时间动态。其变化常常先于转录,比表达更早提示关键调控事件,适合挖掘调控机制和命运决定相关节点 —— 侧重“原因”视角。
建议选择:
- 偏重基因表达/细胞功能变化分析 → 推荐 scRNA-seq
- 注重表观遗传调控或发育早期事件 → 推荐 scATAC-seq
同步分析并整合两类数据的伪时间,有助于更加全面地揭示细胞发育时序与调控机制。
Monocle3 对 scATAC-seq 数据进行拟时序分析的实现
Monocle3 对 scATAC-seq 数据的拟时序分析主要依赖 Cicero 包的扩展。针对单细胞染色质可及性数据的极度稀疏性,Monocle3 采用 LSI(Latent Semantic Indexing)进行预处理,这是 scATAC-seq 分析的核心步骤。
# Seurat 对象转换为 Monocle3 的 CDS 对象
library(SeuratWrappers)
cds <- as.cell_data_set(obj)
# 细胞聚类
cds <- cluster_cells(cds, reduction_method = "UMAP")
# 轨迹学习
cds <- learn_graph(cds, use_partition = TRUE)
# 计算伪时间,指定 root cells 为轨迹起点
# 如未指定 root_cells,则会启动交互式界面选择
cds <- order_cells(cds, reduction_method = "UMAP", root_cells = root_cell_ids)
# 伪时间轨迹可视化
plot_cells(cds, color_cells_by = "pseudotime", show_trajectory_graph = TRUE)关键参数详解与优化建议
- reduction_method:指定降维方法。推荐使用 ATAC 数据对应的 UMAP 降维结果,并确保其名称为 "UMAP"(目前 Monocle3 的
cluster_cells仅支持 UMAP 形式,否则会报错)。 - use_partition = TRUE:是否考虑细胞分区。开启(TRUE)可用于解析存在多个独立发育轨迹的情况,有助于梳理复杂分化路径;如只关注单一路径可设置为 FALSE。
- root_cells:设置轨迹分析的起始细胞,需填写一个或多个 root 细胞的 ID。通常选用发育起点的细胞类型(如干细胞、前体细胞),合理设置 root 细胞有助于确保伪时间方向的生物学意义。
伪时间值提取
分析结束后,可将伪时间值写入 Seurat 对象以便后续分析:
# 添加伪时间信息到 Seurat 对象
obj <- AddMetaData(
object = obj,
metadata = cds@principal_graph_aux@listData$UMAP$pseudotime,
col.name = "Pseudotime"
)
# 可视化伪时间分布
FeaturePlot(obj, "Pseudotime", pt.size = 0.1) +
scale_color_viridis_c()拟时序分析结果展示
伪时间轨迹图
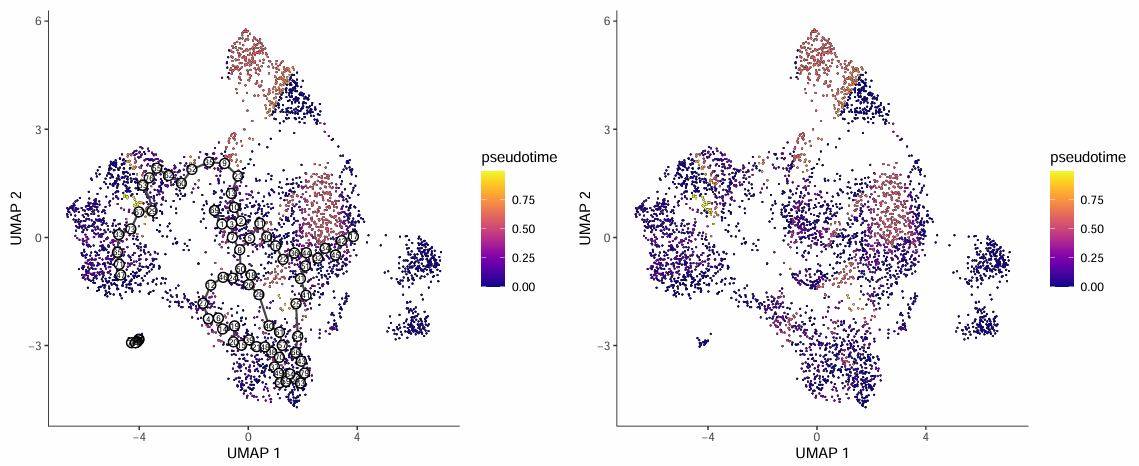
伪时间轨迹图展示了细胞在 UMAP 降维空间中的分布,以及根据伪时间值着色的结果:
- 图中每个点代表一个细胞,黑色线表示拟时序推断得到的轨迹分支路径。
- 细胞点的颜色越深,代表越接近发育起始端;颜色越亮,代表细胞处于发育末端。
- 图中的白底圆圈编号表示轨迹的 root(起始)节点。
- 未与 root 细胞处于同一分区(partition)的离群细胞未被计算伪时间(pseudotime),显示为灰色。
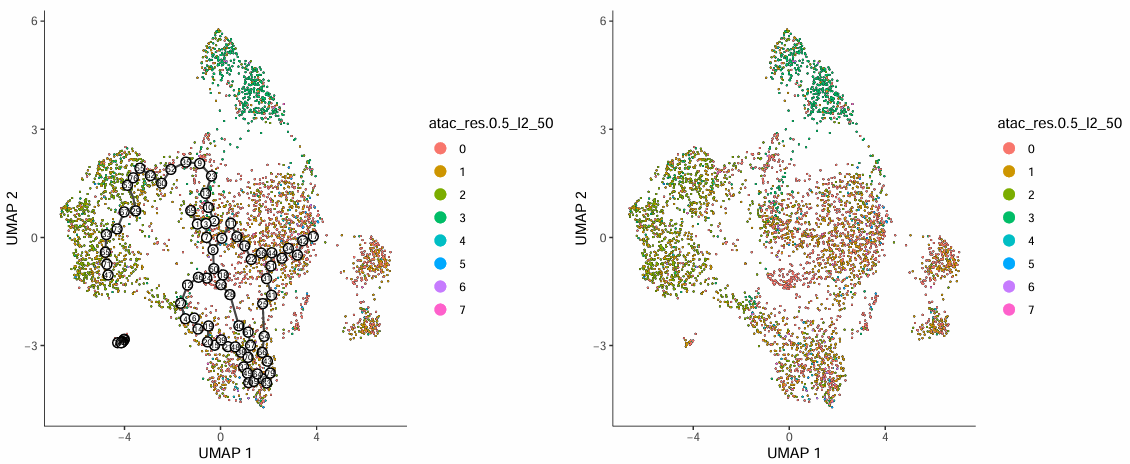
细胞类型轨迹图
该图展示了伪时间值在 UMAP 空间中的分布模式:
- 图中每个点代表一个细胞,不同颜色标识不同的细胞群体,黑色线表示分析得到的轨迹分支路径。
- 黑底圆圈中的数字表示轨迹的分支节点,代表分化方向的分叉位置(可类比为树的枝丫);灰底圆圈表示终末分化状态,对应细胞命运的最终结果(类似树叶)。
- 圆圈内的数字为随机分配,不代表发育顺序高低;分化方向可结合伪时间值(pseudotime)进行判定。
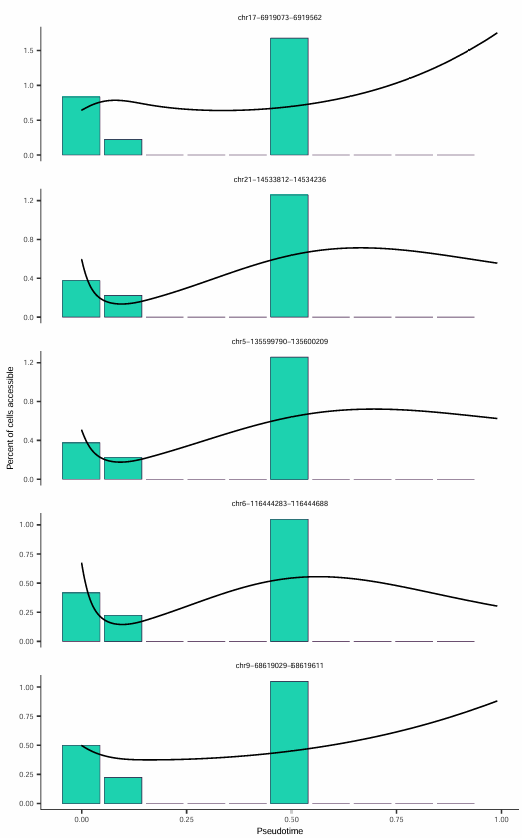
可及性随伪时间变化趋势
随时间可及性增加最显著的五个位点
对于感兴趣的 peaks,可以绘制其可及性沿伪时间的变化曲线:
该图展示了在伪时间轴上可及性增加最为显著的五个染色质区域,每个子图代表一个染色质可及性位点。
x 轴表示伪时间进程,y 轴表示该位点的可及性水平。黑色曲线为平滑拟合线,直观显示了可及性随伪时间变化的整体趋势。
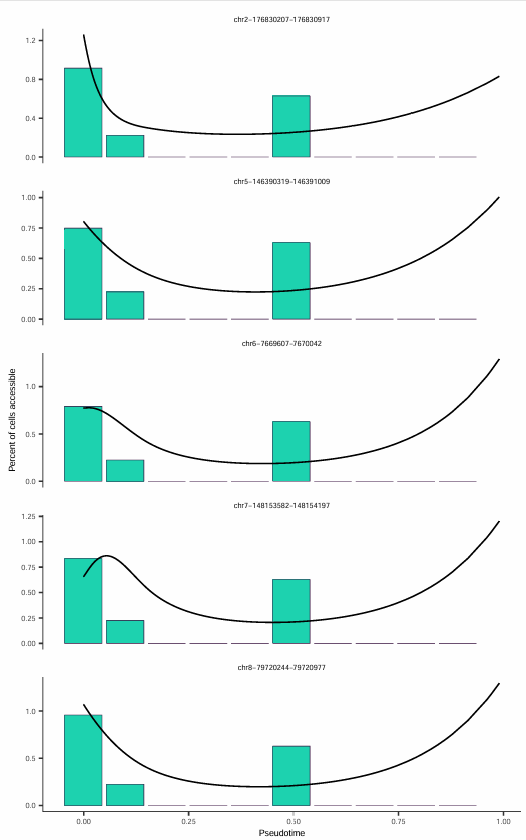
随时间可及性降低最显著的五个位点
通过差异可及性分析,可以识别在伪时间过程中显著变化的 Peaks:
该图展示了在伪时间轴上可及性降低最为显著的五个染色质区域,每个子图代表一个染色质可及性位点。
x 轴表示伪时间进程,y 轴表示该位点的可及性水平,黑色曲线是拟合的平滑线,显示随伪时间变化的总体趋势。
常见问题
Q1:如何选择合适的 root cells?
A: 选择 root cells 是拟时序分析的关键步骤,建议:
- 生物学先验知识:根据已知的生物学知识选择起始细胞类型(如造血干细胞、前体细胞等)
- 轨迹起点识别:在 UMAP 图中,选择位于轨迹分支起点的细胞群体
- 交互式选择:如果不指定
root_cells参数,Monocle3 会启动交互式界面,允许用户手动选择起始细胞 - 多轨迹分析:对于包含多个独立轨迹的数据,需要为每个轨迹分别指定 root cells
Q2:如何处理多个独立的发育轨迹?
A:对于包含多个独立轨迹的数据(如多谱系分化),可以采用以下策略:
- 分区分析:使用
use_partition = TRUE参数,Monocle3 会自动识别不同的细胞分区 - 分别分析:根据细胞类型注释,将数据分为不同的子集,分别进行拟时序分析
- 整合结果:分析完成后,可以将不同轨迹的伪时间值分别添加到 Seurat 对象中,便于比较
Q3:拟时序分析结果如何与其他分析整合?
A: 拟时序分析结果可以与多种分析整合:
- 差异可及性分析:识别沿伪时间变化的 Peaks,揭示动态调控元件
- Motif 分析:对差异 Peaks 进行 Motif 富集,识别关键转录因子
- 多组学整合:与 scRNA-seq 数据整合,揭示染色质可及性与基因表达的时序关联
- 功能注释:结合基因注释,识别受动态调控元件影响的靶基因