单细胞 scATAC-seq & scRNA-seq 双组学标准分析:数据质控
一、质控目的
剔除细胞碎片、双细胞、死细胞等异常barcode,确保分析只包含高质量、真实的单细胞。
油滴包裹单细胞的正确状态
下图为理想情况:一个油滴只包裹一个完整细胞。但实际实验中也可能出现油滴包裹多个细胞(双胞,Doublet),或者油滴内包裹的是一些游离的细胞碎片。两者在质控中都应被去除。
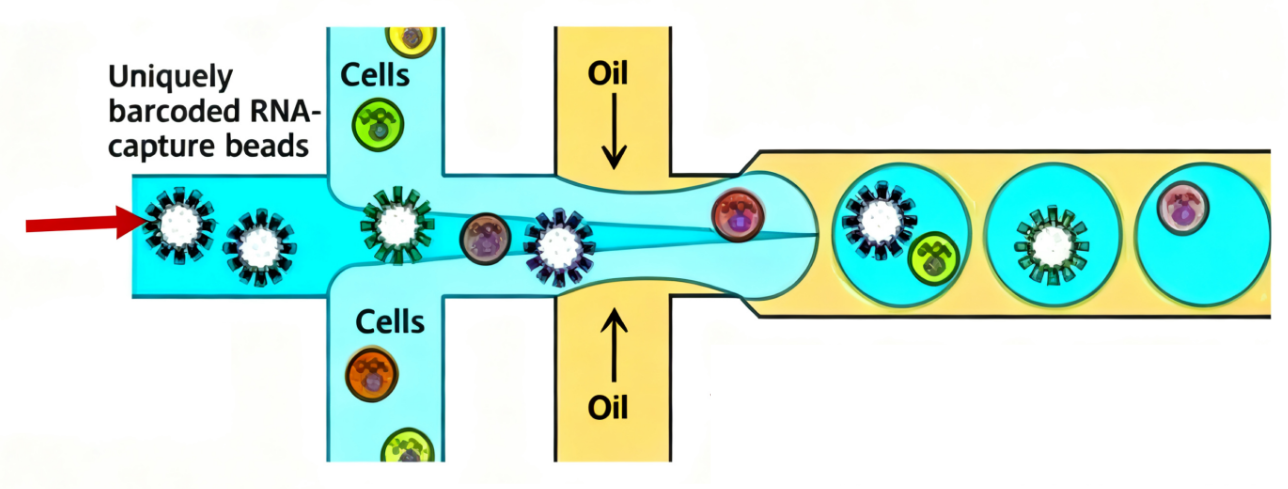
二、关键指标与建议
1) RNA 端质控
核心指标(阈值需按数据灵活调整):
- 每个细胞检测到的基因数(
nFeature_RNA):该指标太低提示是细胞碎片而非真实的细胞,该指标太高提示是双细胞,并非独立的单个细胞,常用阈值建议 200~6000(具体阈值可结合小提琴图适当调整)。 - 每个细胞总 UMI(
nCount_RNA):反映每个细胞内所有转录本分子的总捕获量,通常与基因数协同变化。常用筛选范围为 500–10,000(具体阈值可结合小提琴图适当调整)。 - 线粒体基因占比(
percent.mt):该指标用于过滤可能的死细胞,线粒体占比高于 20% 或 25% 常提示细胞受到应激、破损或处于死亡状态。常用推荐阈值为 ≤10%~20%。
# 计算线粒体基因百分比(以人类基因为例,线粒体基因通常以 "MT-" 开头)
data[["percent.mt"]] <- PercentageFeatureSet(
object = data,
pattern = "^MT-"
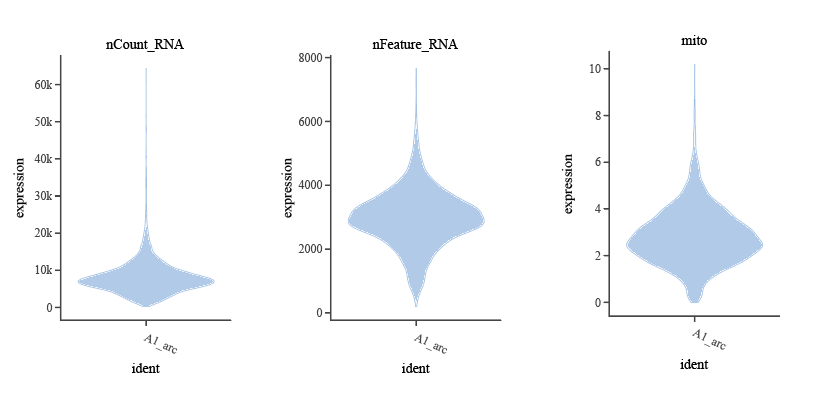
)# 绘制 RNA 端质控小提琴图(nFeature_RNA, nCount_RNA, percent.mt)
VlnPlot(
object = data,
features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3,
pt.size = 0
)小提琴图中离群点(如nFeature_RNA极低或极高,percent.mt异常升高等)往往反映质量不佳的细胞,这些区域适合作为过滤阈值。建议结合经验推荐范围和小提琴图形态,灵活对阈值进行微调,以最大程度保留高质量细胞并去除噪音。
2) ATAC 端质控
建议评估以下指标:
- TSS 富集分数(
TSS.enrichment):衡量细胞染色体状体的一个指标。TSS高富集状态提示染色质开放状态良好,通常对应细胞存活和转录稳态良好。TSS 富集分数过低常见于染色质降解、细胞死亡或技术噪音。可用TSSEnrichment()计算。常用阈值建议 ≥1–2(推荐下限,具体阈值需结合小提琴图适当调整)。
# 运行TSS富集分数计算
data <- TSSEnrichment(
object = data,
assay = "ATAC"
)
# 结果会被添加到 meta.data 的 TSS.enrichment 列- 核小体信号(
nucleosome_signal):描述单核小体片段(~147bp)与无核小体片段(<100bp)的比例。优质细胞由于染色质结构完整,会在片段长度分布中呈现高低交替、约 200bp 除数的周期性波峰,反映细胞内核小体排列和染色质包装的生理状态。核小体信号异常升高提示细胞凋亡、降解或过多双胞等非生理状态。常用推荐阈值 ≤2,大于此值建议剔除。
# 计算核小体信号(nucleosome_signal)
data <- NucleosomeSignal(
object = data,
assay = "ATAC"
)
# 结果会被添加到 meta.data 的 nucleosome_signal 列- ATAC 片段总数(
nCount_ATAC):该指标和上述的nCount_RNA类似,是从ATAC层面来过滤碎片或双细胞的,常见推荐范围为 1,000–80,000(具体阈值需结合小提琴图适当调整)。
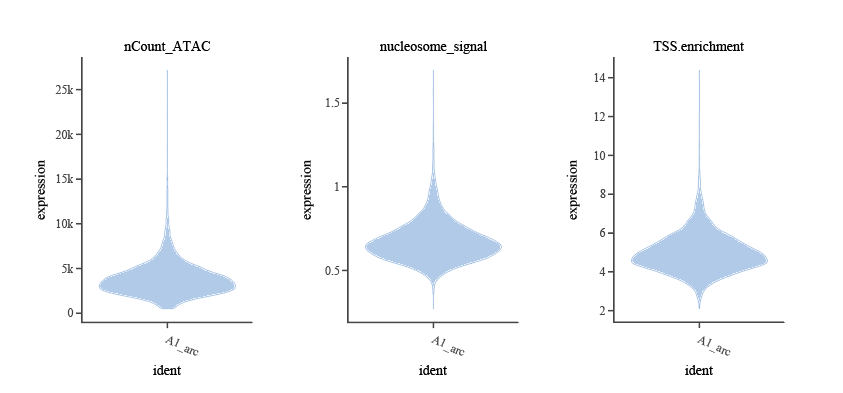
# 绘制 ATAC 端质控小提琴图(TSS.enrichment, nucleosome_signal, nCount_ATAC)
VlnPlot(
object = data,
features = c("TSS.enrichment", "nucleosome_signal", "nCount_ATAC"),
ncol = 3,
pt.size = 0
)同样利用小提琴图帮助我们直观可视化ATAC层面各项 QC 指标的分布,辅助判断合理的过滤阈值,并发现异常或低质量细胞。
三、常见问题
Q1:我用Signac官网教程处理seekARC数据,教程里面有用到一个singlecell.csv文件,你们没提供这个文件给我呀?A:老师用的应该是处理scATAC-seq单组学数据的教程,在单组学教程里面,singlecell.csv文件是为了计算pct_reads_in_peaks指标,从而实现细胞过滤,Signac有提供一胞双组学的指定教程https://stuartlab.org/signac/articles/pbmc_multiomic, 双组学数据过滤是不需要这个指标的,所以我们没有提供。
Q2:为什么在Signac多组学(ATAC+RNA)分析时,不用pct_reads_in_peaks指标来过滤细胞?A:pct_reads_in_peaks(peak区reads占比)主要用于单组学ATAC质量控制,反映信噪比;单组学时没有其他数据支持,需要严格用它剔除低质量细胞。但在多组学数据中,RNA信息可作为ATAC数据质控的互补。如果细胞的RNA数据表现好,即使pct_reads_in_peaks稍低也可能是生物学真实、有价值的细胞。部分亚型细胞天然pct_reads_in_peaks低,若强行筛选可能丢失关键信息。因此,Signac在多组学中更关注TSS.enrichment、nucleosome_signal等稳健指标,通过ATAC与RNA交叉验证整体质量,无需强制用pct_reads_in_peaks过滤。
Q3:中性粒细胞会如何影响多组学数据的质控指标?A: 1)中性粒细胞被激活(如受炎症刺激)时,染色质会解聚并释放到细胞外,形成与组蛋白结合的“中性粒细胞胞外诱捕网”(NETs),以清除病原体。这类事件会产生大量、分布随机的小DNA片段,在测序数据中表现为:
- TSS富集度、overlap peaks等指标显著下降;
- 染色体上这些碎片的占比可高达60%,且大部分难以被归入ATAC调用的peak区域;
- 包含大量此状态中心粒的样本不适合做scATAC-seq测序。
2)未被激活、处于静息(终末分化)状态的中性粒细胞,尽管整体转录活性较低,但其染色质结构完整,各项常规质控指标(如TSS.enrichment、overlap peaks等)通常处于正常范围,仅基因表达量(nFeature_RNA、nCount_RNA)与ATAC片段总量(nCount_ATAC)中位数相对较低。这属于生理现象,实际数据中也常见。