Banksy Spatial Clustering: Principles, Installation, Code Examples, and Interpretation
Load Required R Packages
library(Seurat)
library(Banksy)
library(SummarizedExperiment)
library(SpatialExperiment)
library(scuttle)
library(scater)
library(cowplot)
library(ggplot2)Warning message:
“package ‘SummarizedExperiment’ was built under R version 4.3.2”
Loading required package: MatrixGenerics
Warning message:
“package ‘MatrixGenerics’ was built under R version 4.3.3”
Loading required package: matrixStats
Warning message:
“package ‘matrixStats’ was built under R version 4.3.3”
Attaching package: ‘MatrixGenerics’
The following objects are masked from ‘package:matrixStats’:
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVars
Loading required package: GenomicRanges
Warning message:
“package ‘GenomicRanges’ was built under R version 4.3.3”
Loading required package: stats4
Loading required package: BiocGenerics
Warning message:
“package ‘BiocGenerics’ was built under R version 4.3.2”
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, aperm, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
Position, rank, rbind, Reduce, rownames, sapply, setdiff, sort,
table, tapply, union, unique, unsplit, which.max, which.min
Loading required package: S4Vectors
Warning message:
“package ‘S4Vectors’ was built under R version 4.3.3”
Attaching package: ‘S4Vectors’
The following object is masked from ‘package:utils’:
findMatches
The following objects are masked from ‘package:base’:
expand.grid, I, unname
Loading required package: IRanges
Warning message:
“package ‘IRanges’ was built under R version 4.3.3”
Loading required package: GenomeInfoDb
Warning message:
“package ‘GenomeInfoDb’ was built under R version 4.3.2”
Loading required package: Biobase
Warning message:
“package ‘Biobase’ was built under R version 4.3.3”
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: ‘Biobase’
The following object is masked from ‘package:MatrixGenerics’:
rowMedians
The following objects are masked from ‘package:matrixStats’:
anyMissing, rowMedians
Attaching package: ‘SummarizedExperiment’
The following object is masked from ‘package:SeuratObject’:
Assays
The following object is masked from ‘package:Seurat’:
Assays
Loading required package: SingleCellExperiment
Loading required package: ggplot2
Warning message:
“package ‘ggplot2’ was built under R version 4.3.3”
Warning message:
“package ‘cowplot’ was built under R version 4.3.3”
Read Workflow RDS Data
# Input data here is the Seurat object result file from SeekSpace™ Tools analysis, containing expression matrix and spatial location matrix
obj=readRDS('../../data/AY1739779861133/input.rds')str(obj, max.level = 2)..@ assays :List of 1
..@ meta.data :'data.frame': 32860 obs. of 8 variables:
..@ active.assay: chr "RNA"
..@ active.ident: Factor w/ 16 levels "0","1","2","3",..: 1 2 4 8 3 1 4 4 4 3 ...n .. ..- attr(*, "names")= chr [1:32860] "AAAGGTACGATAACTCGGCACATACAA_1" "AACCACATACAACTCTTGTGATAAGCA_1" "AATGAAGTACCTGACCCCGTGCCGATT_1" "ACGGTCGGCTCTGCCTTTCCTATTAGG_1" ...n ..@ graphs :List of 2
..@ neighbors : list()
..@ reductions :List of 5
..@ images : list()
..@ project.name: chr "SeuratProject"
..@ misc :List of 1
..@ version :Classes 'package_version', 'numeric_version' hidden list of 1
..@ commands :List of 9
..@ tools : list()
# Input here is metadata from SeekSoul™ Online. Banksy analysis doesn't depend on it, but results can be merged into it for SeekSoul™ Online use.
meta=read.table('../../data/AY1739779861133/meta.tsv',header = T, sep = '\t')head(meta)| barcode | orig.ident | nCount_RNA | nFeature_RNA | mito | Sample | raw_Sample | resolution.0.5_d30 | mitorelatedgenes | CellAnnotation | ⋯ | celltype | resolution.1.5_d30 | resolution.1.2_d30 | Celltype | T_sub | Celltype_new | cluster24 | large_celltype | PD1_T | large_celltype_PD1T | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <chr> | <int> | <int> | <dbl> | <chr> | <chr> | <int> | <dbl> | <chr> | ⋯ | <chr> | <int> | <int> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | |
| 1 | AAAGGTACGATAACTCGGCACATACAA_1 | 24110309_T1_T4_T8_expression | 200 | 180 | 5.0 | T8 | 24110309_T1_T4_T8_expression | 0 | 5.0 | B Cell | ⋯ | Neutrophil | 18 | 12 | Macrophage | undefined | B cells | undefined | B_cells | undefined | B_cells |
| 2 | AACCACATACAACTCTTGTGATAAGCA_1 | 24110309_T1_T4_T8_expression | 200 | 175 | 8.0 | T8 | 24110309_T1_T4_T8_expression | 1 | 8.0 | Fibroblast | ⋯ | Fibroblast | 11 | 4 | SMC | undefined | SMC | undefined | SMC | undefined | SMC |
| 3 | AATGAAGTACCTGACCCCGTGCCGATT_1 | 24110309_T1_T4_T8_expression | 200 | 167 | 11.0 | T4 | 24110309_T1_T4_T8_expression | 3 | 11.0 | Tuft Cell | ⋯ | Tuft Cell | 1 | 1 | Epithelial | undefined | Epithelial cells | undefined | Epithelial_cells | undefined | Epithelial_cells |
| 4 | ACGGTCGGCTCTGCCTTTCCTATTAGG_1 | 24110309_T1_T4_T8_expression | 200 | 177 | 1.0 | T1 | 24110309_T1_T4_T8_expression | 7 | 1.0 | T Cell | ⋯ | T_cells | 3 | 2 | T | undefined | T cells | undefined | T_cells | Undefined | T_cells |
| 5 | AGACACTCGAACAGTCTAGACCGCCTT_1 | 24110309_T1_T4_T8_expression | 200 | 185 | 2.0 | T4 | 24110309_T1_T4_T8_expression | 2 | 2.0 | Neutrophil | ⋯ | Neutrophil | 18 | 12 | Macrophage | undefined | B cells | undefined | B_cells | undefined | B_cells |
| 6 | AGGAGGTCGAAATGAGTGCACATACAA_1 | 24110309_T1_T4_T8_expression | 200 | 183 | 9.5 | T8 | 24110309_T1_T4_T8_expression | 0 | 9.5 | B Cell | ⋯ | B_cell | 12 | 8 | B | undefined | B cells | undefined | B_cells | undefined | B_cells |
rownames(meta) = meta$barcode
obj@meta.data = metaPrepare SpatialExperiment Data for Banksy Analysis
colName="Sample"
sample_name="T1"obj_sample = subset(obj,cells = rownames(obj@meta.data[obj@meta.data[colName] == sample_name,]))
sce <- Seurat::as.SingleCellExperiment(obj_sample)
# spatialCoords is the spatial location matrix of cells.
spatialCoords <- obj_sample@reductions$spatial@cell.embeddings
sample_id <- as.character(obj_sample$Sample)
# Convert to SpatialExperiment object, which serves as input for Banksy
spe <- SpatialExperiment(
assays = assays(sce),
rowData = rowData(sce),
colData = colData(sce),
sample_id = sample_id,
spatialCoords = spatialCoords
)Data Preprocessing
# The commented 4 lines below are for filtering, calculating 5% and 98% quantiles of cell UMI counts, retaining cells within this range. Filtering is not applied by default here.
#qcstats <- perCellQCMetrics(spe)
#thres <- quantile(qcstats$total, c(0.05, 0.98))
#keep <- (qcstats$total > thres[1]) & (qcstats$total < thres[2])
#spe <- spe[, keep]
if (dim(spe)[2] <= 50000) {
# Use all genes by default. If using all genes, cell count must be under 60000. Data is normalized to median library size.
spe <- computeLibraryFactors(spe)
aname <- "normcounts"
assay(spe, aname) <- normalizeCounts(spe, log = FALSE)
} else {
# Use highly variable genes
#' Get HVGs
seu <- as.Seurat(spe, data = NULL)
seu <- FindVariableFeatures(seu, nfeatures = 2000)
#' Normalize data
scale_factor <- median(colSums(assay(spe, "counts")))
seu <- NormalizeData(seu, scale.factor = scale_factor, normalization.method = "RC")
#' Add data to SpatialExperiment and subset to HVGs
aname <- "normcounts"
assay(spe, aname) <- GetAssayData(seu)
spe <- spe[VariableFeatures(seu),]
}Perform Banksy Analysis
AGF and k-geometric parameters determine which surrounding values are used to calculate the neighborhood, using normalized expression (normcounts in se object) to calculate the neighborhood feature matrix. For SeekSpace data, these parameters have relatively small impact; set agf to TRUE and k_geom to c(15, 30).
- AGF Parameter
To describe the neighborhood, Banksy calculates weighted neighborhood mean (H_0) and azimuthal Gabor filter (H_1), the latter for estimating gene expression gradients. Setting compute_agf to TRUE computes both H_0 and H_1.
- k-geometric Parameter
k_geom specifies the number of neighbors for calculating each H_m (m = 0,1). If a single value is specified, each feature matrix uses the same k_geom. Alternatively, multiple k_geom values can be provided for each feature matrix. Here, we use k_geom[1]=15 and k_geom[2]=30 for H_0 and H_1 respectively. More neighbors are used for gradient calculation.
For datasets generated using Visium v1/v2, use k_geom = 18 (if compute_agf = TRUE, use k_geom <- c(18, 18)), as this corresponds to using two concentric rings of points around each point as the neighborhood.
k_geom <- c(15, 30)
spe <- Banksy::computeBanksy(spe, assay_name = aname, compute_agf = TRUE, k_geom = k_geom)“sparse->dense coercion: allocating vector of size 1.7 GiB”
Computing neighbors...n
Spatial mode is kNN_median
Parameters: k_geom=15
Done
Computing neighbors...n
Spatial mode is kNN_median
Parameters: k_geom=30
Done
Computing harmonic m = 0
Using 15 neighbors
Done
Computing harmonic m = 1
Using 30 neighbors
Centering
Done
- lambda Parameter
Lambda is a mixing parameter in [0,1] determining how much spatial information is included, significantly affecting results. "lambda = 0" corresponds to non-spatial clustering; small "lambda" runs Banksy in cell typing mode; high "lambda" runs Banksy in domain discovery mode. Our goal is domain segmentation, so we generally use larger lambda values. For SeekSpace data, 0.6 or 0.8 yields expected segmentation. In rare cases, if results are irregular with many small regions, try increasing lambda; if regions are too large/coarse, try decreasing it.
Here lambda is set to c(0.6, 0.8), so results for both 0.6 and 0.8 will be calculated and appear in the final cluster results.
lambda <- c(0.6, 0.8)
npcs = 30
set.seed(1000)
spe <- Banksy::runBanksyPCA(spe, use_agf = TRUE, lambda = lambda, npcs = npcs)
spe <- Banksy::runBanksyUMAP(spe, use_agf = TRUE, lambda = lambda, npcs = npcs)“sparse->dense coercion: allocating vector of size 1.7 GiB”
Warning message in asMethod(object):
“sparse->dense coercion: allocating vector of size 1.7 GiB”
- resolution Parameter
This parameter works like Seurat's res parameter; higher values mean more clusters. Here c(0.4, 0.6) means results for resolution 0.4 and 0.6 will be calculated and appear in the final cluster results.
- npcs Parameter
Specifies the number of principal components. Default is 20. If there are many domains, try setting npcs to 50 to increase the final cluster count.
- algo Parameter
The algo parameter can be leiden, louvain, kmeans, or mclust.
Leiden and louvain are similar, automatically calculating cluster count, requiring resolution (higher = more clusters). Both yield very "regular" segment domains, with default leiden being the best. Kmeans and mclust are similar, requiring manual cluster count specification. Kmeans segment domains are quite accurate in both mouse brain and human eye data. In summary, start with the default leiden algorithm. If adjusting lambda, npcs, and resolution doesn't yield expected segment domains, try kmeans.
resolution <- c(0.2,0.4)
spe <- Banksy::clusterBanksy(spe, use_agf = TRUE, lambda = lambda, algo = 'leiden', npcs = npcs, resolution = resolution)When algo is kmeans, use the code below
# If using kmeans clustering, replace the clusterBanksy line above with the commented line below; no other code changes needed.
# se <- Banksy::clusterBanksy(se, use_agf = TRUE, lambda = lambda, algo = "kmeans", kmeans.centers = c(5,10,15,20,25))# To visually compare clustering results, use connectClusters to relabel. Run as default, no modification needed.
spe <- Banksy::connectClusters(spe)clust_M1_lam0.8_k50_res0.4 --> clust_M1_lam0.8_k50_res0.2
clust_M1_lam0.6_k50_res0.4 --> clust_M1_lam0.8_k50_res0.4
colnames(colData(spe))- 'barcode'
- 'orig.ident'
- 'nCount_RNA'
- 'nFeature_RNA'
- 'mito'
- 'Sample'
- 'raw_Sample'
- 'resolution.0.5_d30'
- 'mitorelatedgenes'
- 'CellAnnotation'
- 'intestine'
- 'Group'
- 'FCN1_LGR5'
- 'PCDH17_LGR5'
- 'CD31_LGR5'
- 'CLDN2_P'
- 'LGR5_ALL_CELL'
- 'LGR5_ALL'
- 'LGR5_ALL_1'
- 'PCDH17_ALL'
- 'PCDH17_LGR5_ALL'
- 'CLDN2_P1'
- 'FCN1_LGR5_2'
- 'LGR5_TOTAL'
- 'CLXCL6_epi'
- 'CLXCL6_epi_1'
- 'resolution.1.0_d30'
- 'celltype'
- 'resolution.1.5_d30'
- 'resolution.1.2_d30'
- 'Celltype'
- 'T_sub'
- 'Celltype_new'
- 'large_celltype'
- 'PD1_T'
- 'large_celltype_PD1T'
- 'ident'
- 'sample_id'
- 'sizeFactor'
- 'clust_M1_lam0.6_k50_res0.2'
- 'clust_M1_lam0.6_k50_res0.4'
- 'clust_M1_lam0.8_k50_res0.2'
- 'clust_M1_lam0.8_k50_res0.4'
Result Visualization
Explanation of cluster labels in the image below, taking "clust_M1_lam0.6_k50_res0.4" as an example:
clust: indicates cluster result
M1: indicates use_agf = TRUE
lam0.6: indicates lambda=0.6
k50: indicates k_neighbors=50, default value, no modification needed
res0.4: indicates resolution=0.4
# Spatial visualization of all clustering labels from Banksy parameter combinations
cnames <- grep("^clust", colnames(colData(spe)), value = TRUE)
colData(spe) <- cbind(colData(spe), spatialCoords(spe))
plots <- list()
for (i in seq_along(cnames)) {
plots[[i]] <- plotColData(spe, x = "spatial_1", y = "spatial_2", point_size = 1, colour_by = cnames[i]) + coord_equal()
}
options(repr.plot.height=16, repr.plot.width=16)
plot_grid(plotlist = plots, ncol = 2)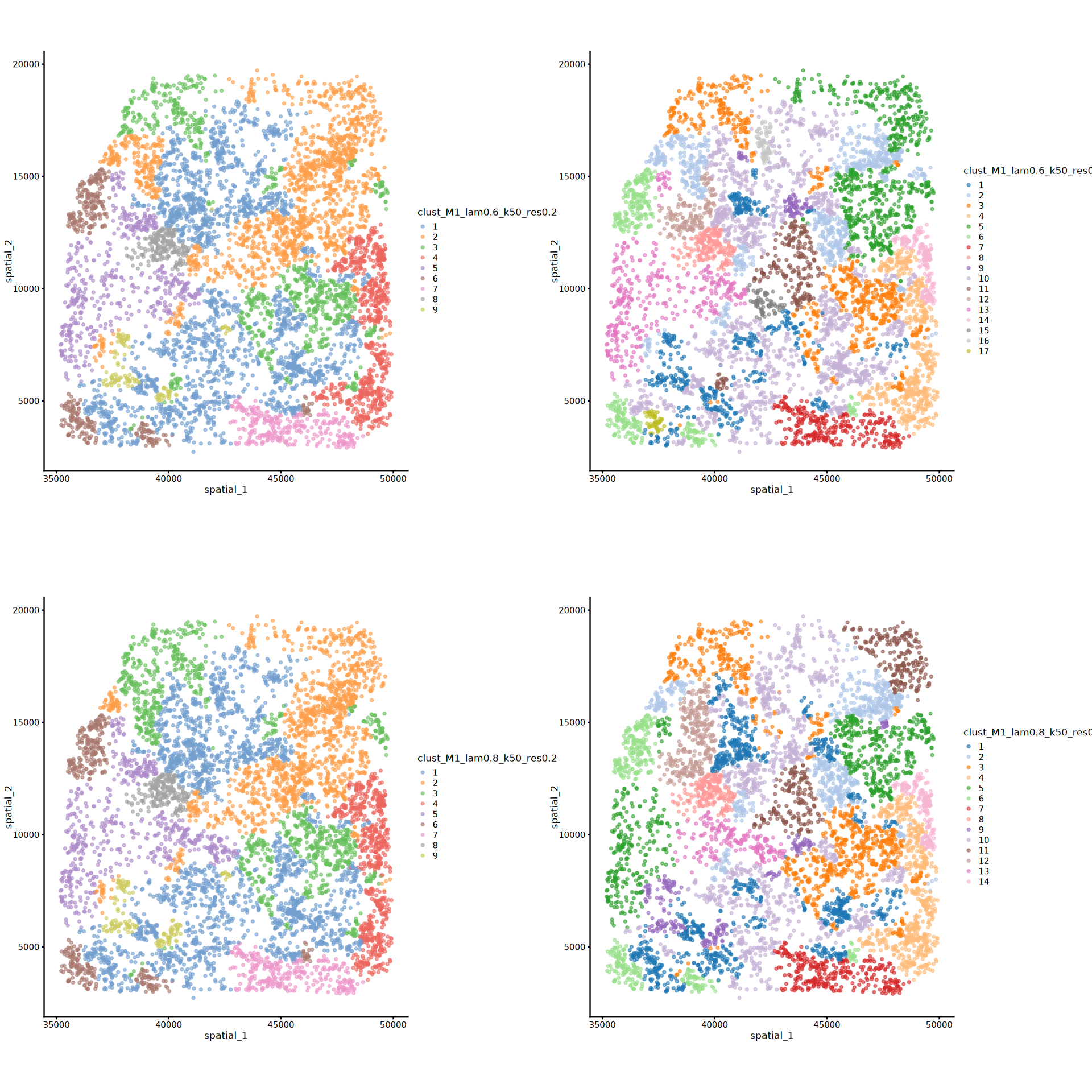
# UMAP visualization of all clustering labels from Banksy parameter combinations
rdnames <- grep("UMAP.*lam", reducedDimNames(spe), value = TRUE)
umap_plots <- list()
plot_num = 1
for (i in seq_along(rdnames)) {
cnnames_use = cnames[which(grepl(gsub('UMAP_', '', rdnames[i]), cnames))]
for (j in seq_along(cnnames_use)){
umap_plots[[plot_num]] <- plotReducedDim(spe, dimred = rdnames[i], colour_by = cnnames_use[j]) + coord_equal()
plot_num = plot_num + 1
}
}
options(repr.plot.height=16, repr.plot.width=16)
plot_grid(plotlist = umap_plots, ncol = 2)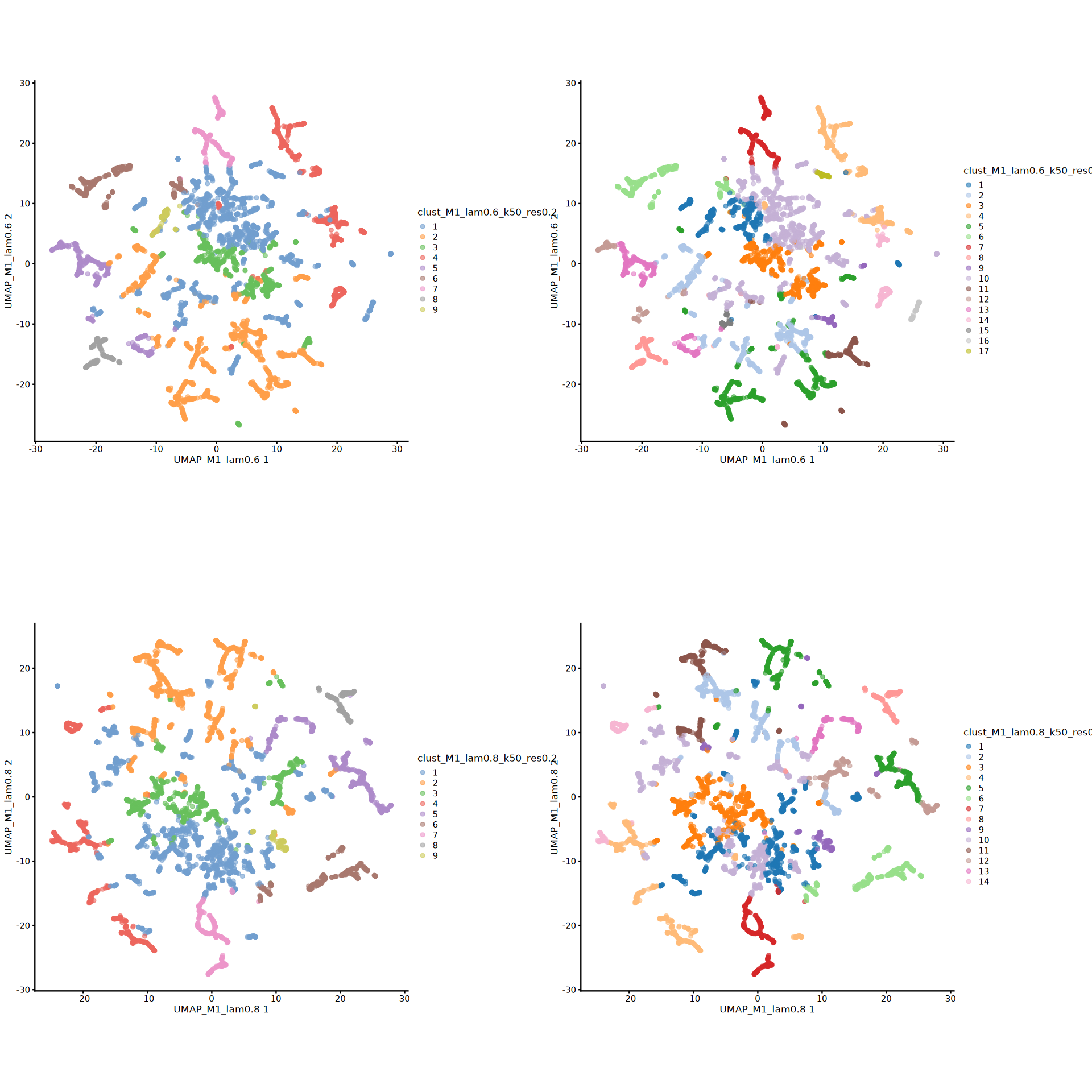
Output Results
# Save Banksy analysis results
save(spe, file='banksy.rda')# Save Banksy clustering results, which can be merged into Seurat metadata
write.csv(colData(spe), 'banksy_colData.csv')