scMethyl + RNA Multi-omics: Integration Analysis Tutorial Based on MOFA+ Algorithm
Module Introduction
This tutorial details how to use the MOFA+ (Multi-Omics Factor Analysis v2) framework for multi-omics integration analysis of single-cell methylation (DNA Methylation) and transcriptome (RNA) data.
MOFA+ Analysis Principles
MOFA+ is an unsupervised statistical framework designed to infer latent low-dimensional factors (Latent Factors) from multi-omics data. Its core idea is to decompose data from different omics modalities (Views) into a set of shared or private latent factors.
- Input Data: Matrices containing multiple omics modalities (such as RNA and DNA Methylation), where rows represent aligned cells and columns represent their respective features (genes or methylation regions).
- Latent Factors: The model extracts low-dimensional factors driving data variation through matrix factorization.
- Shared Factors: Capture biological variation co-existing in multiple omics modalities (such as cell type, cell cycle, etc.), explaining the variance of both RNA and methylation data simultaneously.
- Private Factors: Capture variation existing only in a single omics modality (such as technical noise specific to one omics, batch effects, or specific biological signals).
- Model Reconstruction: The data is decomposed into the product of a Factor Matrix and a Weight Matrix. The weight matrix measures the contribution of each feature to a specific factor.
- Sparsity: MOFA+ introduces sparsity priors, so each factor is associated with only a small number of key features (genes or methylation regions). This regularization not only prevents overfitting but also significantly improves the biological interpretability of the model.
Analysis Objectives
- Multi-omics Dimensionality Reduction: Compress high-dimensional RNA and methylation data into a small number of latent factors, retaining the main biological variation.
- Variance Explanation: Quantify and assess the contribution of each factor to the overall variance of different omics modalities (Variance Explained).
- Joint Clustering: Construct a neighbor graph based on the extracted low-dimensional factor space for more robust joint dimensionality reduction (UMAP) and cell clustering (Leiden).
Preparation
Import Dependencies and Define Helper Functions
This section primarily completes the following tasks:
- Import Analysis Libraries: Including standard single-cell analysis libraries (
scanpy,muon), methylation analysis library (ALLCools), and plotting library (matplotlib). - Define Data Loading and Processing Functions:
mcds_to_adata: Convert MCDS format (methylation storage format) toAnnDataobject.mcds_get_mc_frac_adata: Read and process Methylation Fraction matrix, supporting blacklist region filtering and highly variable feature calculation.compute_cg_ch_level: Calculate global CG and CH methylation levels for each cell.check_barcode_overlap: Check cell Barcode overlap between RNA and Methylation data to ensure multi-omics data comes from the same batch of cells.parse_gtf: Parse GTF annotation file to extract gene type information.
import os
# Set environment variables to ensure the correct conda environment and binaries are used
os.environ['PATH'] = "/PROJ/home/weiqiuxia/micromamba/envs/muon_env/bin:/opt/conda/bin:/opt/conda/condabin:/usr/local/sbin:/usr/local/bin:/usr/sbin:/usr/bin:/sbin:/bin:/PROJ2/FLOAT/public/bin"
r_base_path = '/PROJ/home/weiqiuxia/micromamba/envs/muon_env'
os.environ['R_HOME'] = os.path.join(r_base_path, 'lib', 'R')
# Set shared library path
os.environ['LD_LIBRARY_PATH'] = os.path.join(r_base_path, 'lib', 'R', 'lib') + ':' + os.environ.get('LD_LIBRARY_PATH', '')
# Set R library paths
r_lib_path = os.path.join(r_base_path, 'lib', 'R', 'library')
os.environ['R_LIBS'] = r_lib_path
os.environ['R_LIBS_USER'] = r_lib_path
os.environ['R_LIBS_SITE'] = r_lib_path
import re
import glob
from ALLCools.mcds import MCDS
from ALLCools.clustering import tsne, significant_pc_test, log_scale, lsi, binarize_matrix, filter_regions, cluster_enriched_features, ConsensusClustering, Dendrogram, get_pc_centers
from ALLCools.clustering.doublets import MethylScrublet
from ALLCools.plot import *
import scanpy as sc
import scanpy.external as sce
from harmonypy import run_harmony
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
from matplotlib.lines import Line2D
import warnings
import xarray as xr
from ALLCools.clustering import one_vs_rest_dmg
from scipy import sparse
import muon as mu
from muon import atac as ac
from skmisc.loess import loess
import mofax as mfx
import re
from collections import defaultdict
sc.settings.verbosity = 0
warnings.filterwarnings('ignore')
try:
from matplotlib_venn import venn2
HAS_VENN = True
except Exception:
HAS_VENN = False
plt.rcParams['figure.dpi'] = 80
plt.rcParams['savefig.dpi'] = 300
plt.rcParams['figure.figsize'] = (5,5)def mcds_to_adata(mcds_path, var_dim = 'chrom20k', mc_type = 'CGN', quant_type = 'hypo-score'):
'''
Convert the hypo-score matrix to an AnnData object
'''
# Read MCDS file
mcds = MCDS.open(mcds_path, obs_dim = 'cell', var_dim = var_dim)
# Get the score matrix and convert to AnnData
adata = mcds.get_score_adata(mc_type=mc_type, quant_type=quant_type, sparse = True)
return(adata)
def mcds_get_mc_frac_adata(mcds_path, var_dim = 'chrom20k', mc_type = 'CGN', obs_dim = 'cell',
min_cov = 1, met_matrix_type = 'mc_frac', hvf_method='SVR' ,n_top_feature = 2000,
clip_norm_value = 10,
black_list_path = None, ):
'''
Convert the methylation fraction matrix or M-value matrix to an AnnData object
'''
# 1. Open MCDS dataset
mcds = MCDS.open(
mcds_path,
obs_dim = 'cell',
var_dim = var_dim
)
# 2. Compute mean coverage per feature (Coverage Mean)
if "mc_type" in mcds[f'{var_dim}_da'].dims:
feature_cov_mean = mcds.sel(count_type="cov").sum(dim="mc_type").mean(dim=obs_dim).squeeze().to_pandas()
else:
feature_cov_mean = mcds[f'{var_dim}_da'].sel(count_type="cov").mean(dim=obs_dim).squeeze().to_pandas()
mcds.coords[f'{var_dim}_cov_mean'] = (var_dim, feature_cov_mean)
# Filter out low-coverage features
mcds = mcds.filter_feature_by_cov_mean(min_cov=min_cov)
# 3. (Optional) Remove blacklist regions (Blacklist Regions)
if black_list_path:
mcds = mcds.remove_black_list_region(
black_list_path = black_list_path,
f = 0.2
)
load = True
obs_dim = 'cell'
# 4. Compute methylation matrix (M-value or Methylation Fraction)
if met_matrix_type == 'mvalue':
mcds.add_m_value(var_dim=var_dim)
da_suffix = 'mvalue'
else:
if met_matrix_type == 'mc_frac':
mcds.add_mc_frac(var_dim=var_dim,clip_norm_value=clip_norm_value,normalize_per_cell=True)
else:
clip_norm_value = None
mcds.add_mc_frac(var_dim=var_dim,clip_norm_value=clip_norm_value,normalize_per_cell=False)
da_suffix = 'frac'
# Load data into memory
if load and (mcds.get_index(obs_dim).size < 20000):
mcds[f'{var_dim}_da_{da_suffix}'].load()
# 5. Compute highly variable features (Highly Variable Features)
if hvf_method == 'SVR':
mcg_hvf = mcds.calculate_hvf_svr(
var_dim = var_dim,
mc_type = mc_type,
da_suffix = da_suffix,
n_top_feature = n_top_feature,
plot = False)
else:
mcg_hvf = mcds.calculate_hvf(
var_dim = var_dim,
mc_type = mc_type,
da_suffix = da_suffix,
n_top_feature = n_top_feature,
plot = False)
# 6. Create AnnData object and merge HVF metadata
mcg_adata = mcds.get_adata(
mc_type=mc_type,
var_dim = var_dim,
select_hvf = True,
da_suffix = da_suffix)
mcg_adata.var = mcg_adata.var.merge(mcg_hvf, right_index = True, left_index = True)
return mcg_adata
def compute_cg_ch_level(df):
'''
Compute CG and CH methylation levels
'''
# Select coverage (cov) and methylation (mc) columns for CG and CH contexts
cg_cov_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_cov')]
cg_mc_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_mc')]
ch_cov_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_cov')]
ch_mc_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_mc')]
# Compute global CG methylation fraction
df['CpG_cov'] = df[cg_cov_col].sum(axis=1)
df['CpG_mc'] = df[cg_mc_col].sum(axis=1)
df['CpG%'] = round(df['CpG_mc'] * 100 / df['CpG_cov'], 2)
# Compute global CH methylation fraction
df['CH_cov'] = df[ch_cov_col].sum(axis=1)
df['CH_mc'] = df[ch_mc_col].sum(axis=1)
df['CH%'] = round(df['CH_mc'] * 100 / df['CH_cov'], 2)
return df
def check_barcode_overlap(rna_adata, met_adata, barcode_map = None):
'''
Check barcode overlap between transcriptome and methylation data, and reset RNA barcodes to the corresponding methylation barcodes.
'''
# Compute barcode overlap rate
rna_adata.obs['original_barcode'] = [ re.sub('_.*', '', i) for i in rna_adata.obs.index ]
met_adata.obs['original_barcode'] = [ re.sub('_.*', '', i) for i in met_adata.obs.index ]
overlap_rate = len(set(rna_adata.obs['original_barcode']) & set(met_adata.obs['original_barcode'])) / met_adata.shape[0]
# If overlap rate is low (<10%), try converting barcodes using the mapping table
if overlap_rate < 0.1:
met2rna_barcode = barcode_map[barcode_map['m_cb'].isin(met_adata.obs['original_barcode'])]
rna_adata = rna_adata[rna_adata.obs['original_barcode'].obs.index.isin(met2rna_barcode['gex_cb'])]
met2rna_barcode = met2rna_barcode.set_index('gex_cb')
rna_adata.obs['gex_cb'] = rna_adata.obs.index
rna_adata.obs.index = met2rna_barcode.loc[rna_adata.obs['original_barcode'],'m_cb']
# Re-check overlap rate
overlap_rate_test = len(set(rna_adata.obs.index) & set(met_adata.obs.index)) / met_adata.shape[0]
chemistry = 'DD-MET3'
if overlap_rate_test < 0.1:
raise Exception('The overlap rate between RNA data and MET data barcode is less than 10%')
else:
chemistry = 'DD-MET5'
rna_adata.obs.index = rna_adata.obs['original_barcode']
met_adata.obs.index = met_adata.obs['original_barcode']
# Keep only cells shared by both modalities
common_index = list(set(rna_adata.obs.index) & set(met_adata.obs.index))
rna_adata = rna_adata[rna_adata.obs.index.isin(common_index)]
rna_adata.obs['chemistry'] = chemistry
met_adata = met_adata[met_adata.obs.index.isin(common_index)]
met_adata.obs['chemistry'] = chemistry
return rna_adata, met_adata, chemistry
def parse_gtf(gtf_file, output_file):
'''
Read GTF, get gene_type information for genes.
'''
with open(gtf_file, 'r') as f_in, open(output_file, 'w') as f_out:
for line in f_in:
if line.startswith('#'):
continue
parts = line.strip().split('\t')
if len(parts) < 9:
continue
feature_type = parts[2]
if feature_type != 'gene':
continue
attributes = parts[8]
attr_dict = {}
# Basic parsing of GTF attributes: key "value"; key "value";
for attr in attributes.split(';'):
attr = attr.strip()
if not attr:
continue
if ' ' in attr:
key, value = attr.split(' ', 1)
value = value.strip('"')
attr_dict[key] = value
gene_id = attr_dict.get('gene_id', '')
gene_name = attr_dict.get('gene_name', '')
gene_type = attr_dict.get('gene_type', '')
# Construct the first column
if gene_id and gene_name:
col1 = f"{gene_id}_{gene_name}"
elif gene_id:
col1 = gene_id
else:
col1 = gene_name # Should not happen based on GTF spec but good to handle
f_out.write(f"{col1}\t{gene_type}\n")
def rds_to_matrix(filepath, outdir):
import rpy2.robjects as robjects
robjects.globalenv['rdsfile'] = filepath
robjects.globalenv['outdir'] = outdir
print(f"R_HOME: {os.environ['R_HOME']}")
print(f"R_LIBS: {os.environ['R_LIBS']}")
print(f"LD_LIBRARY_PATH: {os.environ['LD_LIBRARY_PATH']}")
# Run R code and retrieve results
robjects.r('''
library(Seurat)
library(qs)
if(endsWith(rdsfile, ".rds")){
obj <- readRDS(rdsfile)
}else{
obj <- qread(rdsfile)
}
dir.create(outdir, recursive = TRUE)
Matrix::writeMM(GetAssayData(obj, assay = "RNA", layer = "counts"), file = file.path(outdir, "matrix.mtx") )
cmd <- paste0("gzip ", file.path(outdir, "matrix.mtx"))
system(cmd)
features <- data.frame(Feature_id = rownames(obj), Feature_name = rownames(obj), Feature_type = "Gene Expression")
write.table(features, file.path(outdir, "features.tsv"), row.names = F, col.names = F, sep = "\t", quote = F)
cmd <- paste0("gzip ", file.path(outdir, "features.tsv"))
system(cmd)
barcodes <- data.frame(Barcode = colnames(obj))
write.table(barcodes, file.path(outdir, "barcodes.tsv"), row.names = F, col.names = F, sep = "\t", quote = F)
cmd <- paste0("gzip ", file.path(outdir, "barcodes.tsv"))
system(cmd)
meta <- obj@meta.data
write.table(tibble::rownames_to_column(meta, "barcode"), file.path(outdir, "meta.csv"), row.names = FALSE, col.names = TRUE, sep = ",", quote = F)
''')Input Directory Structure Description
Important Note: This workflow has specific requirements for the input directory structure. The program will automatically retrieve RNA and methylation-related input files based on the indir parameter. It is recommended to organize the data as follows:
data/AY1768874914782/ (indir)
├── input.rds # Transcriptome data (Seurat object or qs format)
└── methylation # Methylation data root directory
└── demoWTJW969-task-1 # Task/Batch name
└── WTJW969 # Sample name
├── allcools_generate_datasets
│ └── WTJW969.mcds # MCDS dataset folder
│ ├── chrom100k
│ ├── chrom10k
│ ├── chrom20k # Methylation score matrix for 20kb windows
│ └── ...
└── split_bams
├── filtered_barcode_reads_counts.csv # Filtered Barcode mapped reads counts
└── WTJW969_cells.csv # Filtered cell QC information (CpG count, coverage, etc.)- RNA Data Retrieval: Match
.rdsor.qsfiles under theindirpath. - Methylation Data Retrieval: Retrieve MCDS files and associated QC CSV files under the
indir/methylationpath.
# indir: input directory
indir = '/home/weiqiuxia/workspace/data/AY1768874914782/'
# outdir: output directory for results
outdir = '/home/weiqiuxia/workspace/project/weiqiuxia/MOFA_results'
# dd_met3_barcode_map: barcode mapping table for DD-MET3
dd_met3_barcode_map = '/home/weiqiuxia/workspace/project/weiqiuxia/DD-M_bUCB3_whitelist.csv'
# black_list_path: regions to filter (BED). Regions with 20% overlap will be removed from methylation features
black_list_path = '/home/weiqiuxia/workspace/project/weiqiuxia/hg38-blacklist-sort.v2.bed'
# resolution: clustering resolution for methylation (float > 0)
resolution = 1
# anno_col: cell annotation column name in RNA metadata
anno_col = 'CellAnnotation'
# sample_col: sample column name (default `Sample`); should match sample names under `indir`
sample_col = 'Sample'
# rna_n_top_feature: number of highly variable genes for RNA
rna_n_top_feature = 2000
# matrix_type: methylation matrix type. Options: hypo-score | mvalue
met_matrix_type = 'mvalue'
# var_dim: methylation feature dimension(s). Multiple values are allowed
var_dim = ['geneslop2k']
# total_cpg_number_cutoff: CpG count cutoff
total_cpg_number_cutoff = 500
# mc_type: methylation type: CGN. Currently only CGN is supported
mc_type = 'CGN'
# min_cov: filter out features with mean coverage below this value
min_cov = 10
# normalize_per_cell: whether to use posterior estimation for methylation fraction. True denoises/implicates; False uses raw methylation levels
normalize_per_cell=True
# met_n_top_feature: method for selecting highly variable features. Possible values: SVR | Based
hvf_method='SVR'
# met_n_top_feature: number of highly variable features to select for methylation
met_n_top_feature = 2500
# total_cpg_number_cutoff: CpG count cutoff
total_cpg_number_cutoff = 500os.makedirs(outdir, exist_ok=True)# Read DD-MET3 barcode mapping table
dd_met3_barcode_map_df = pd.read_csv(dd_met3_barcode_map, header = 0)# First locate the input .qs/.rds file, convert it to a matrix, then load with scanpy
for i in os.listdir(indir):
if i.endswith('.qs'):
rds_to_matrix(os.path.join(indir, i), os.path.join(outdir, 'input_matrix'))
breakR_LIBS: /PROJ/home/weiqiuxia/micromamba/envs/muon_env/lib/R/library
LD_LIBRARY_PATH: /PROJ/home/weiqiuxia/micromamba/envs/muon_env/lib/R/lib:
# Load RNA data with scanpy
adata_rna = sc.read_10x_mtx(f'{outdir}/input_matrix/')
meta = pd.read_csv(f'{outdir}/input_matrix/meta.csv', index_col = 0, header = 0)
adata_rna.obs = meta
adata_rna.obs['CellAnnotation'] = adata_rna.obs['CellAnnotation'].astype('category')Methylation Data Preprocessing
Single-cell methylation data is highly sparse and has distinct signal distribution characteristics, so specific preprocessing strategies are needed for the specified methylation matrix type (met_matrix_type) to ensure downstream analysis accuracy.
We support the following two common data types and their corresponding processing workflows:
Hypo-score Matrix:
- Binarization: Use
binarize_matrixfunction to convert continuous Hypo-score to binary signals. We consider sites withhypo-score > 0.95as significantly hypomethylated (labeled as 1), and the rest as background (labeled as 0). This helps reduce technical noise and highlight biological signals. - TF-IDF Transformation: Apply TF-IDF (Term Frequency-Inverse Document Frequency) algorithm. This is a standard method for handling sparse binary data (similar to scATAC-seq analysis), effectively correcting sequencing depth differences and reducing the weight of ubiquitous features to extract cell-specific information.
- Binarization: Use
M-value Matrix:
- Logit Transformation: Based on raw Methylation Fraction, calculate M-value using the formula
np.log2((frac + alpha) / (1 - frac + alpha)). This transformation maps the [0, 1] methylation rate interval to the real number domain, providing better variance stability, making it more suitable for linear models and dimensionality reduction analysis.
- Logit Transformation: Based on raw Methylation Fraction, calculate M-value using the formula
Subsequently, we will perform Feature Selection based on the processed matrix, screening for methylation regions with the largest variation across cells, to prepare for subsequent multi-modal integration.
# Load methylation data and take the intersection with transcriptome data to ensure cells and barcodes match
keep_col = ['barcode', 'total_cpg_number', 'genome_cov', 'reads_counts', 'CpG%', 'CH%']
obj_met_list = defaultdict(defaultdict)
obj_rna_list = defaultdict()
indirs = indir.split(',')
samples = []
mcds_paths = {}
sample_path = {}
analysis_batch_dir = os.listdir(os.path.join(indir, 'methylation'))
for i in analysis_batch_dir:
if os.path.isdir(os.path.join(indir, 'methylation', i)):
a_batch_dir = os.path.join(indir, 'methylation', i)
# Get all samples under the analysis batch
samples.extend([ i for i in os.listdir(a_batch_dir)])
for s in samples:
mcds_path = os.path.join(a_batch_dir, f'{s}','allcools_generate_datasets', f'{s}.mcds')
mcds_paths[s] = mcds_path
sample_path[s] = a_batch_dir
# Read methylation QC results
s_cells_meta = pd.read_csv(os.path.join(a_batch_dir, s, 'split_bams', f'{s}_cells.csv'), header = 0)
s_cells_meta['barcode'] = [b.replace('_allc.gz','') for b in s_cells_meta['cell_barcode']]
# Read mapped reads per cell
s_reads_counts = pd.read_csv(os.path.join(a_batch_dir, s, 'split_bams', 'filtered_barcode_reads_counts.csv'), header = 0)
s_merged_df = s_cells_meta.merge(s_reads_counts, how = 'inner', left_on = 'barcode', right_on = 'barcode')
# Merge the dataframes above
s_merged_df = compute_cg_ch_level(s_merged_df)
s_merged_df = s_merged_df[keep_col]
s_merged_df = s_merged_df.set_index('barcode')
# Read methylation matrix and build AnnData
if met_matrix_type == 'hypo-score':
# Handle hypo-score data
if isinstance(var_dim, list):
for vd in var_dim:
# Convert MCDS to AnnData
obj = mcds_to_adata(
mcds_path,
var_dim = vd)
# Merge metadata
obj.obs = obj.obs.merge(s_merged_df, left_index = True, right_index = True)
# Filter low-quality cells
obj = obj[obj.obs['total_cpg_number'] >= total_cpg_number_cutoff ]
obj.obs['Sample'] = s
# Check whether RNA and methylation barcodes match within the same sample; take intersection
adata_rna_s = adata_rna[adata_rna.obs['orig.ident'] == s]
obj_rna_list[s], obj, chemistry = check_barcode_overlap(adata_rna_s, obj, dd_met3_barcode_map_df)
obj_met_list[vd][s] = obj
else:
obj = mcds_to_adata(
mcds_path,
var_dim = var_dim)
obj.obs = obj.obs.merge(s_merged_df, left_index = True, right_index = True)
obj = obj[obj.obs['total_cpg_number'] >= total_cpg_number_cutoff ]
obj.obs['Sample'] = s
adata_rna_s = adata_rna[adata_rna.obs['orig.ident'] == s]
obj_rna_list[s], obj, chemistry = check_barcode_overlap(adata_rna_s, obj, dd_met3_barcode_map_df)
obj_met_list[var_dim][s] = obj
else:
# Handle mc_frac or mvalue data
if isinstance(var_dim, list):
for vd in var_dim:
obj = mcds_get_mc_frac_adata(
mcds_path,
var_dim = vd,
mc_type = mc_type,
min_cov = min_cov,
met_matrix_type = met_matrix_type,
hvf_method=hvf_method ,
n_top_feature = met_n_top_feature,
black_list_path = black_list_path
)
obj.obs = obj.obs.merge(s_merged_df, left_index = True, right_index = True)
obj = obj[obj.obs['total_cpg_number'] >= total_cpg_number_cutoff ]
obj.obs['Sample'] = s
adata_rna_s = adata_rna[adata_rna.obs['orig.ident'] == s]
obj_rna_list[s], obj, chemistry = check_barcode_overlap(adata_rna_s, obj, dd_met3_barcode_map_df)
obj_met_list[vd][s] = obj
else:
obj = mcds_get_mc_frac_adata(
mcds_path,
var_dim = var_dim,
mc_type = mc_type,
min_cov = min_cov,
met_matrix_type = met_matrix_type,
hvf_method=hvf_method ,
n_top_feature = met_n_top_feature,
black_list_path = black_list_path
)
obj.obs = obj.obs.merge(s_merged_df, left_index = True, right_index = True)
obj = obj[obj.obs['total_cpg_number'] >= total_cpg_number_cutoff ]
obj.obs['Sample'] = s
adata_rna_s = adata_rna[adata_rna.obs['orig.ident'] == s]
obj_rna_list[s], obj, chemistry = check_barcode_overlap(adata_rna_s, obj, dd_met3_barcode_map_df)
obj_met_list[var_dim][s] = obj
else:
raise Exception('file not found!')After cov mean filter: 25666 geneslop2k 66.5%
261 geneslop2k features removed due to overlapping (bedtools intersect -f 0.2) with black list regions.
Fitting SVR with gamma 0.0417, predicting feature dispersion using mc_frac_mean and cov_mean.
Total Feature Number: 25405
Highly Variable Feature: 2500 (9.8%)
select_hvf is True, but no highly variable feature results found, use all features instead.
# If there are multiple samples, concatenate the matrices
if len(obj_rna_list) > 1:
adata_rna = obj_rna_list[samples[0]].concatenate([obj_rna_list[i] for i in samples[1:] ])
else:
adata_rna = obj_rna_list[samples[0]]
del obj_rna_list
adata_met_list = {}
for i in obj_met_list.keys():
if len(obj_met_list[i]) > 1:
adata_met_list[i] = obj_met_list[i][samples[0]].concatenate([obj_met_list[i][s] for s in samples[1:] ])
else:
adata_met_list[i] = obj_met_list[i][samples[0]].copy()
del obj_met_listfor i in adata_met_list.keys():
if met_matrix_type == 'hypo-score':
# 1. Binarization: convert continuous hypo-score to a binary signal (0/1), cutoff=0.95
binarize_matrix(adata_met_list[i], cutoff=0.95)
# 2. Region filtering: remove low-quality or uninformative genomic regions
filter_regions(adata_met_list[i])
# Save raw data to layers
adata_met_list[i].layers['raw'] = adata_met_list[i].X.copy()
# 3. TF-IDF normalization: correct sequencing depth and emphasize modality-specific signal (scale_factor=1e4)
ac.pp.tfidf(adata_met_list[i], scale_factor=1e4)
# 4. Feature selection: select highly variable methylation regions (Highly Variable Features)
sc.pp.highly_variable_genes(adata_met_list[i], flavor='seurat_v3', n_top_genes=met_n_top_feature, batch_key='Sample')
# 5. LSI dimensionality reduction: SVD-based method suitable for sparse data
ac.tl.lsi(adata_met_list[i])
else:
# Processing logic for non hypo-score data
min_samples = 2 if adata_met_list[i].obs[sample_col].value_counts().shape[0] > 1 else 1
feature_select_cols = [col for col in adata_met_list[i].var.columns if col.startswith('feature_select')]
adata_met_list[i].var['highly_variable'] = adata_met_list[i].var[feature_select_cols].sum(axis=1) >= min_samplesRNA Data Preprocessing
RNA data preprocessing follows the standard Scanpy workflow, aiming to remove noise and preserve biological signals:
- Quality Control (QC):
sc.pp.filter_cells: Filter out low-quality cells with too few detected genes.sc.pp.filter_genes: Filter out genes expressed in very few cells.
- Normalization:
sc.pp.normalize_total: Eliminate technical effects caused by sequencing depth (library size) differences, typically scaling each cell to 10,000 reads.
- Log Transformation:
sc.pp.log1p: Performtransformation. This makes the data distribution closer to normal, stabilizes variance, and compresses the dynamic range of expression levels.
- Feature Selection:
sc.pp.highly_variable_genes: Select highly variable genes (HVGs) with significant biological variation. These genes contain the main information distinguishing different cell types and are used for subsequent dimensionality reduction analysis.
# Mark mitochondrial genes (Mitochondrial genes)
# Use "MT-" for human, and "Mt-" for mouse
adata_rna.var["mt"] = adata_rna.var_names.str.startswith("MT-")
# Mark ribosomal genes (Ribosomal genes)
adata_rna.var["ribo"] = adata_rna.var_names.str.startswith(("RPS", "RPL"))
# Mark hemoglobin genes (Hemoglobin genes)
adata_rna.var["hb"] = adata_rna.var_names.str.contains("^HB[^(P)]")
# Compute QC metrics (Quality Control Metrics)
# qc_vars: gene sets for which to compute percentages (e.g. mitochondrial fraction)
sc.pp.calculate_qc_metrics(adata_rna, qc_vars=["mt", "ribo", "hb"], inplace=True, log1p=True)# 1. Backup raw count data
adata_rna.layers["counts"] = adata_rna.X.copy()
# 2. Total-count normalization (Normalization)
# Normalize each cell's library size to the median to remove sequencing depth effects
sc.pp.normalize_total(adata_rna)
# 3. Log transformation (Log transformation)
# Apply log(x+1) to make the distribution closer to normal and reduce skewness
sc.pp.log1p(adata_rna)
# 4. Select highly variable genes (Highly Variable Genes)
# Identify genes with the highest variability across cells for downstream dimensionality reduction and clustering
sc.pp.highly_variable_genes(adata_rna, flavor='seurat_v3', n_top_genes=2000, batch_key="Sample")Here, we previously performed separate batch correction, dimensionality reduction, and clustering annotation on the transcriptome data. This step aims to establish a Single-modality Baseline, to subsequently compare with the MOFA+ multi-modal integration analysis results and assess whether introducing methylation data improves cell clustering clarity or biological interpretability.
#sc.pp.scale(adata_rna, max_value=10)
sc.tl.pca(adata_rna)if len(samples) > 1:
ho = run_harmony(
adata_rna.obsm['X_pca'],
meta_data=adata_rna.obs,
vars_use='Sample',
random_state=0,
max_iter_harmony=20)
adata_rna.obsm['X_pca_harmony'] = ho.Z_corr
rna_reduc_use = 'X_pca_harmony'
else:
rna_reduc_use = 'X_pca'
sc.pp.neighbors(adata_rna,use_rep = rna_reduc_use, random_state = 20)
sc.tl.umap(adata_rna, random_state = 20)
sc.tl.tsne(adata_rna,use_rep=rna_reduc_use, random_state = 20)
sc.tl.leiden(adata_rna, resolution=resolution, random_state=20)Figure Legend: This figure shows the UMAP plot of cell clustering based on transcriptome data.
- Left Panel: Each dot represents a cell, and colors represent different cell clusters (Leiden).
- Right Panel: Each dot represents a cell, and colors represent different samples (Sample).
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi=80)
sc.pl.umap(adata_rna, color = ['leiden','Sample'], wspace=0.3)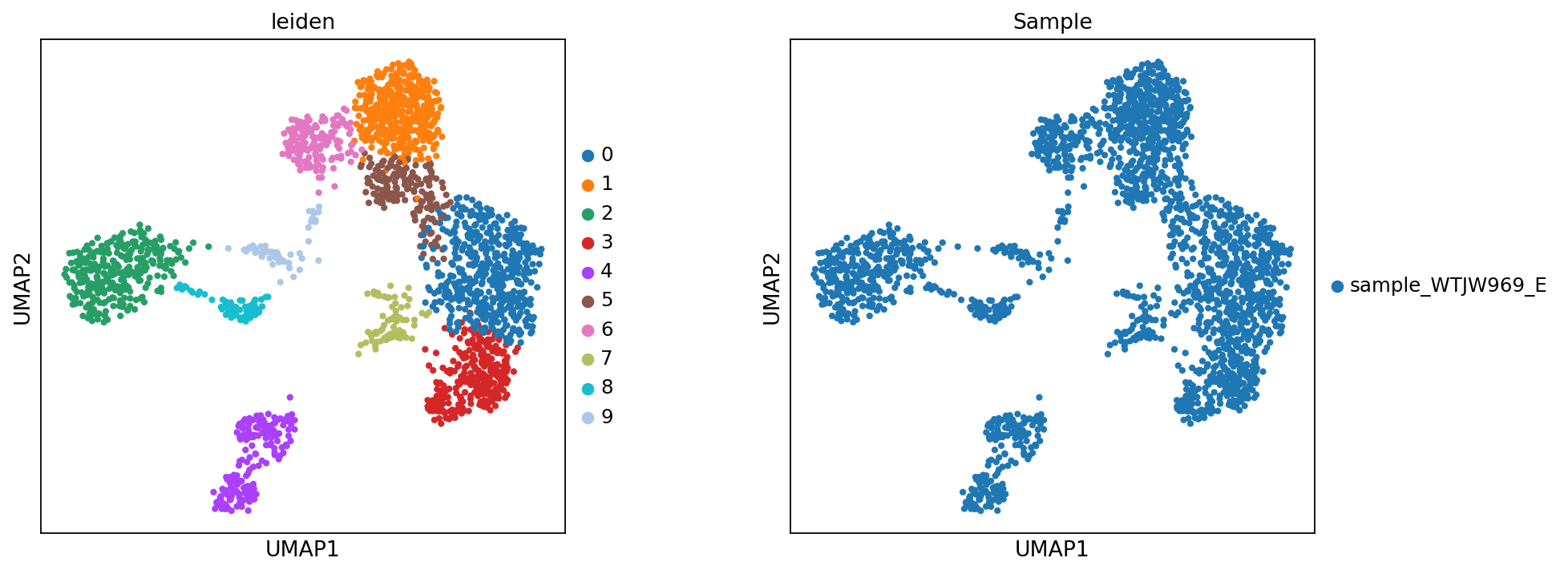
Figure Legend: This figure shows the expression distribution of specific marker genes in the UMAP dimensionality reduction space of RNA data.
- X and Y axes: The two main dimensions after UMAP reduction (UMAP1 and UMAP2).
- Color: Color depth indicates expression levels of marker genes; darker means higher expression. Markers shown include:
CD3D,THEMIS(T cells),NKG7(NK cells),CD8A(CD8+ T cells),CD4(CD4+ T cells),CD79AandMS4A1(B cells),JCHAIN(Plasma cells),LYZ(Monocytes),LILRA4(pDC cells).- Dots: Each dot represents an individual cell.
- Purpose: Through marker gene expression patterns, different cell types can be preliminarily identified and validated to assist in cell type annotation.
colors = ["#F5E6CC", "#FDBB84", "#E34A33", "#7F0000"]
my_warm_cmap = mcolors.LinearSegmentedColormap.from_list("white_orange_red", colors, N=256)
marker_names = ["CD3D", "THEMIS", "NKG7", "CD8A", "CD4", "IL7R", "S100A4", "CCR7",
"CD79A","MS4A1","JCHAIN",
"LYZ","FCN1","FCGR3A","CST3","MKI67"]
sc.pl.umap(
adata_rna,
color=marker_names,
ncols=4,
color_map=my_warm_cmap,
vmax='p99',
s=10,
frameon=False,
vmin=0,
show=False
)
plt.gcf().set_size_inches(12, 8)
plt.show()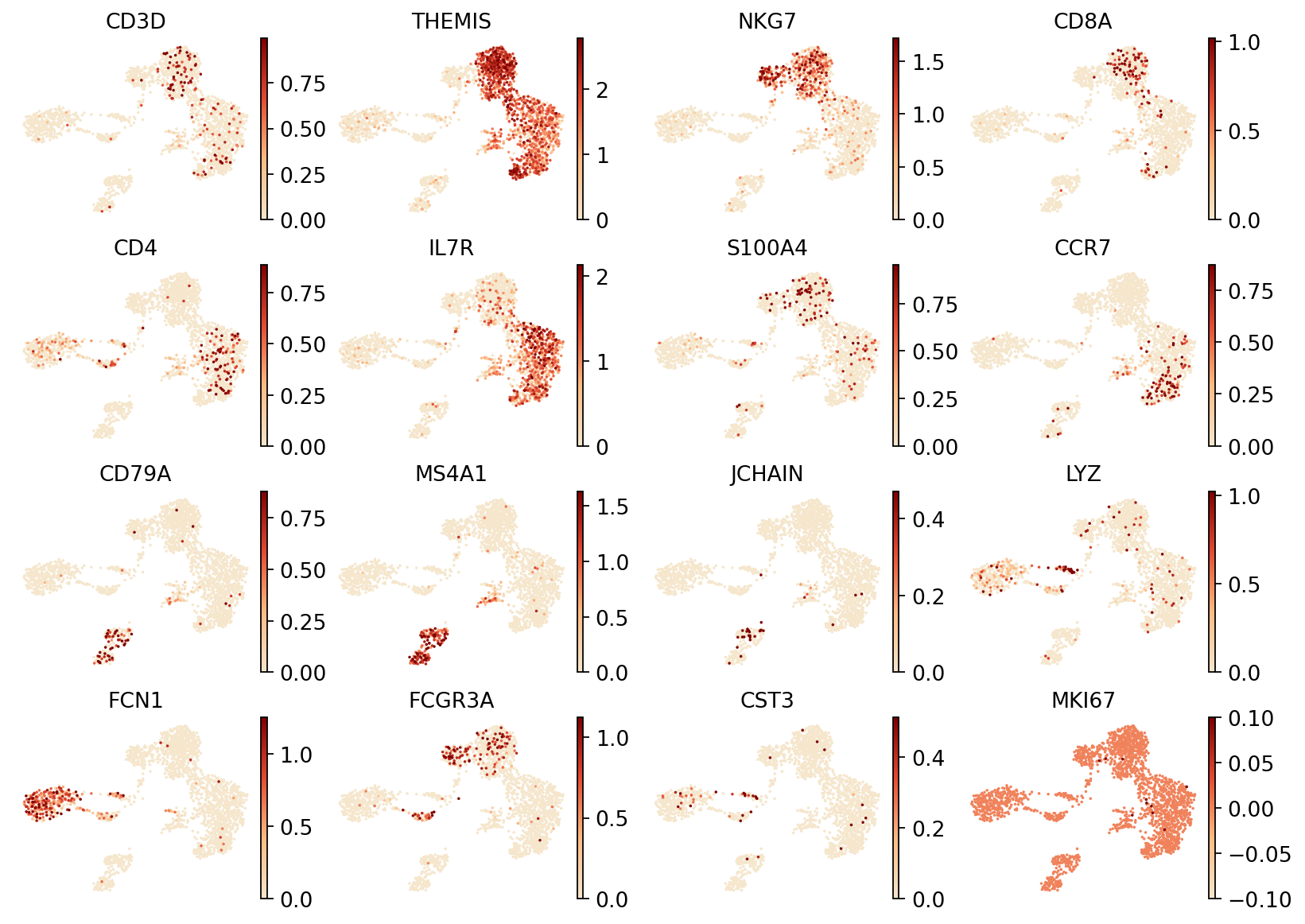
celltype = {
'0': 'Navie CD4+ T',
'1': 'CD8+ T',
'2': 'CD14+ Mono',
'3': 'Memory CD4+ T',
'4': 'B',
'5': 'T',
'6': 'NK',
'7': 'Multiplet',
'8': 'FCGR3A+ Mono',
'9': 'DC'
}
adata_rna.obs['celltype'] = adata_rna.obs['leiden'].map(celltype)if not 'celltype' in adata_rna.obs.columns:
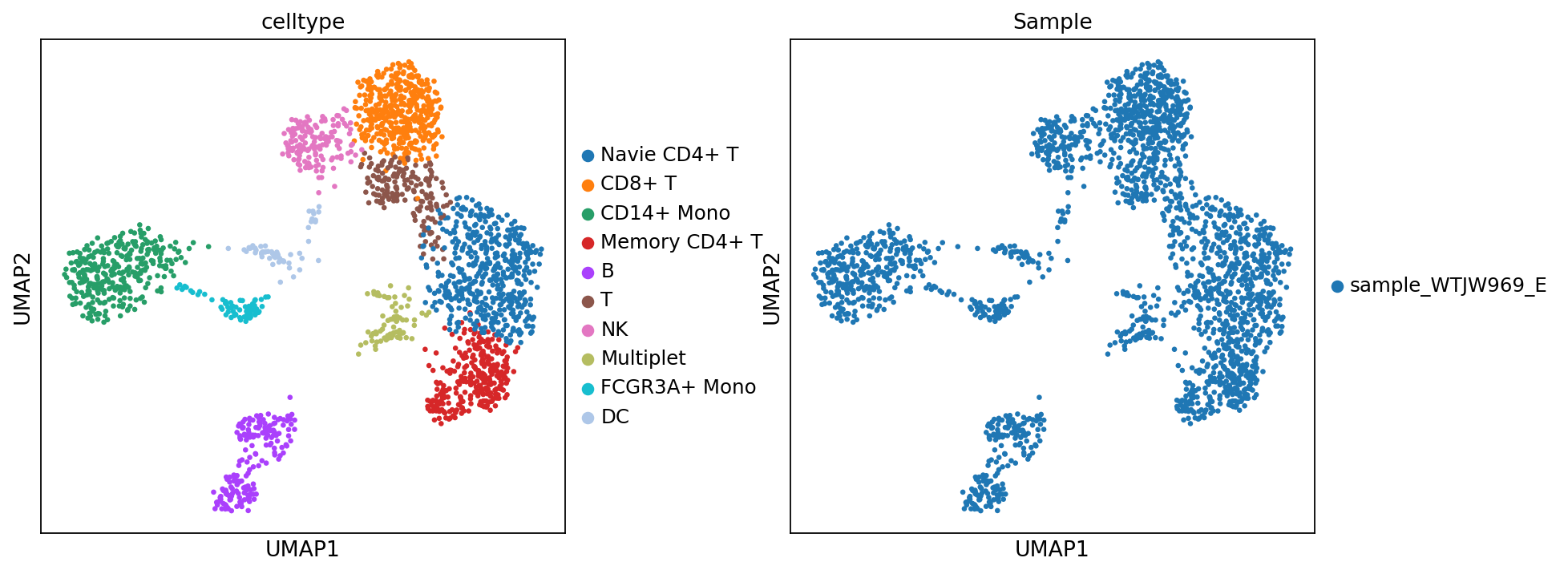
adata_rna.obs['celltype'] = adata_rna.obs['leiden']Figure Legend: This figure shows the UMAP plot of cell type based on transcriptome data.
- Left Panel: Each dot represents a cell, and colors represent different cell type (celltype).
- Right Panel: Each dot represents a cell, and colors represent different samples (Sample).
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi = 80)
sc.pl.umap(
adata_rna,
color=['celltype', 'Sample'],
ncols=2,
s=35,
wspace=0.3
)
Build MuData Object and Train MOFA+ Model
os.makedirs(f"{outdir}/models", exist_ok=True)mu_list = {}
mu_list['rna'] = adata_rna
for i in adata_met_list.keys():
mu_list[f'methy_{i}'] = adata_met_list[i]
mu_listobs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'mito', 'Sample', 'raw_Sample', 'resolution.0.5_d30', 'CellAnnotation', 'original_barcode', 'chemistry', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'leiden', 'celltype'
var: 'gene_ids', 'feature_types', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne', 'leiden', 'leiden_colors', 'Sample_colors', 'celltype_colors'
obsm: 'X_pca', 'X_umap', 'X_tsne'
varm: 'PCs'
layers: 'counts'
obsp: 'distances', 'connectivities',
'methy_geneslop2k': AnnData object with n_obs × n_vars = 2189 × 25405
obs: 'total_cpg_number', 'genome_cov', 'reads_counts', 'CpG%', 'CH%', 'Sample', 'original_barcode', 'chemistry'
var: 'chrom', 'end', 'start', 'cov_mean', 'mean', 'dispersion', 'cov', 'score', 'feature_select', 'highly_variable'}
Build Multi-modal Container (MuData) and Model Training
MuData is a data structure designed specifically for multi-omics data, capable of storing and managing data from multiple modalities (e.g., RNA, Methylation) and their associations simultaneously.
MOFA+ Model Training Parameters Description:
- groups_label: Used for multi-sample/multi-batch analysis, the model will attempt to correct batch effects.
- n_factors: Expected number of latent factors to discover (default 10). More factors can capture more details but increase computational load and may introduce noise.
- scale_views: Whether to scale each modality. Default is
Falseto balance data scale differences between modalities. - convergence_mode: Convergence mode, with
slow,medium, andfastoptions.slowmode trains more thoroughly for more stable results;fastmode is quicker.
# Build a MuData object to integrate RNA and methylation data
mdata = mu.MuData(mu_list)
mdata.obs['Sample'] = mdata['rna'].obs['Sample']
mdata.obs['celltype'] = mdata['rna'].obs['celltype']
mdata.obs['CpG%'] = mdata[mdata.mod_names[1]].obs['CpG%']
mdataMuData object with n_obs × n_vars = 2189 × 64011
obs: 'Sample', 'celltype', 'CpG%'
var: 'highly_variable'
2 modalities
rna: 2189 x 38606
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'mito', 'Sample', 'raw_Sample', 'resolution.0.5_d30', 'CellAnnotation', 'original_barcode', 'chemistry', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'leiden', 'celltype'
var: 'gene_ids', 'feature_types', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne', 'leiden', 'leiden_colors', 'Sample_colors', 'celltype_colors'
obsm: 'X_pca', 'X_umap', 'X_tsne'
varm: 'PCs'
layers: 'counts'
obsp: 'distances', 'connectivities'
methy_geneslop2k: 2189 x 25405
obs: 'total_cpg_number', 'genome_cov', 'reads_counts', 'CpG%', 'CH%', 'Sample', 'original_barcode', 'chemistry'
var: 'chrom', 'end', 'start', 'cov_mean', 'mean', 'dispersion', 'cov', 'score', 'feature_select', 'highly_variable'# Intersect cells across modalities to ensure one-to-one correspondence
mu.pp.intersect_obs(mdata)
# Run the MOFA+ model
mu.tl.mofa(mdata,
use_raw = False,
groups_label = 'Sample', # Sample grouping column for handling batch effects
scale_views=True, # Balance weights across modalities
n_factors=10, # Number of latent factors
convergence_mode='slow', # Use slow mode for more robust convergence
use_var = 'highly_variable', # Train using highly variable features only
outfile=f"{outdir}/models/pbmc_969_multi_methy.hdf5") # Model output path #########################################################
### __ __ ____ ______ ###
### | \/ |/ __ \| ____/\ _ ###
### | \ / | | | | |__ / \ _| |_ ###
### | |\/| | | | | __/ /\ \_ _| ###
### | | | | |__| | | / ____ \|_| ###
### |_| |_|\____/|_|/_/ \_\ ###
### ###
#########################################################
Scaling views to unit variance...
Loaded view='rna' group='sample_WTJW969_E' with N=2189 samples and D=2000 features...
Loaded view='methy_geneslop2k' group='sample_WTJW969_E' with N=2189 samples and D=2500 features...
Model options:
- Automatic Relevance Determination prior on the factors: True
- Automatic Relevance Determination prior on the weights: True
- Spike-and-slab prior on the factors: False
- Spike-and-slab prior on the weights: True
Likelihoods:
- View 0 (rna): gaussian
- View 1 (methy_geneslop2k): gaussian
######################################
## Training the model with seed 1 ##
######################################
Converged!
#######################
## Training finished ##
#######################
Warning: Output file /home/weiqiuxia/workspace/project/weiqiuxia/MOFA_results/models/pbmc_969_multi_methy.hdf5 already exists, it will be replaced
Saving model in /home/weiqiuxia/workspace/project/weiqiuxia/MOFA_results/models/pbmc_969_multi_methy.hdf5...
Saved MOFA embeddings in .obsm['X_mofa'] slot and their loadings in .varm['LFs'].
mdata.uns = mdata.uns or dict()MOFA Factor Visualization
One of the core results of MOFA+ factor analysis is the inferred latent factors. We can visualize the distribution of these factors in cells to understand biological differences between different cell types.
Figure Legend: This figure (scatter plot matrix) shows the distribution of the first 10 MOFA factors (pairwise) in cells.
- Color: Represents cell type (
rna:celltype) or methylation level (methy_geneslop2k:CpG%).- Observation: If a factor (e.g., Factor 1) can clearly separate a certain cell type (e.g., B cells) from other cells, it indicates that this factor captures specific biological signals of that cell type.
mu.pl.mofa(mdata, color="celltype", components=["1,2", "3,4", "5,6", "7,8", "9,10"])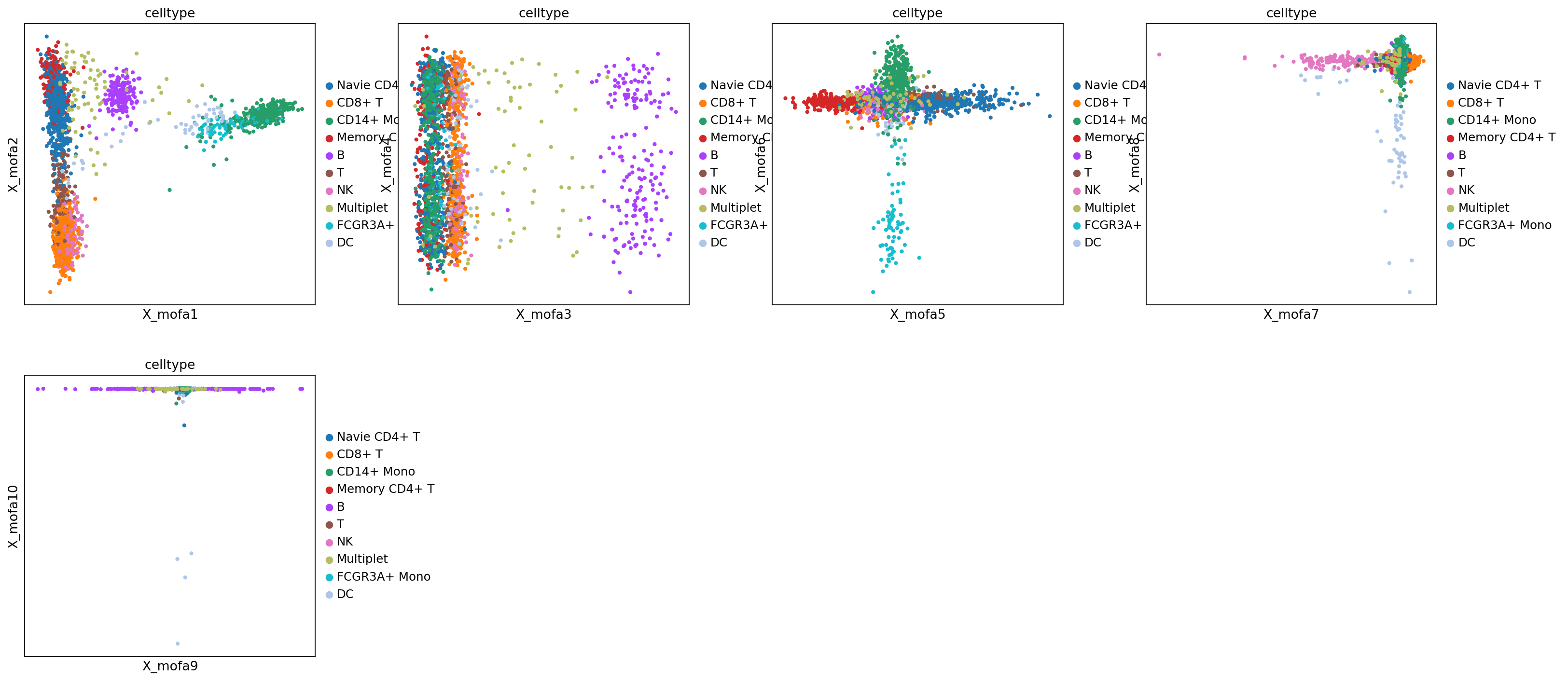
mu.pl.mofa(mdata, color="CpG%", components=["1,2", "3,4", "5,6", "7,8","9,10"])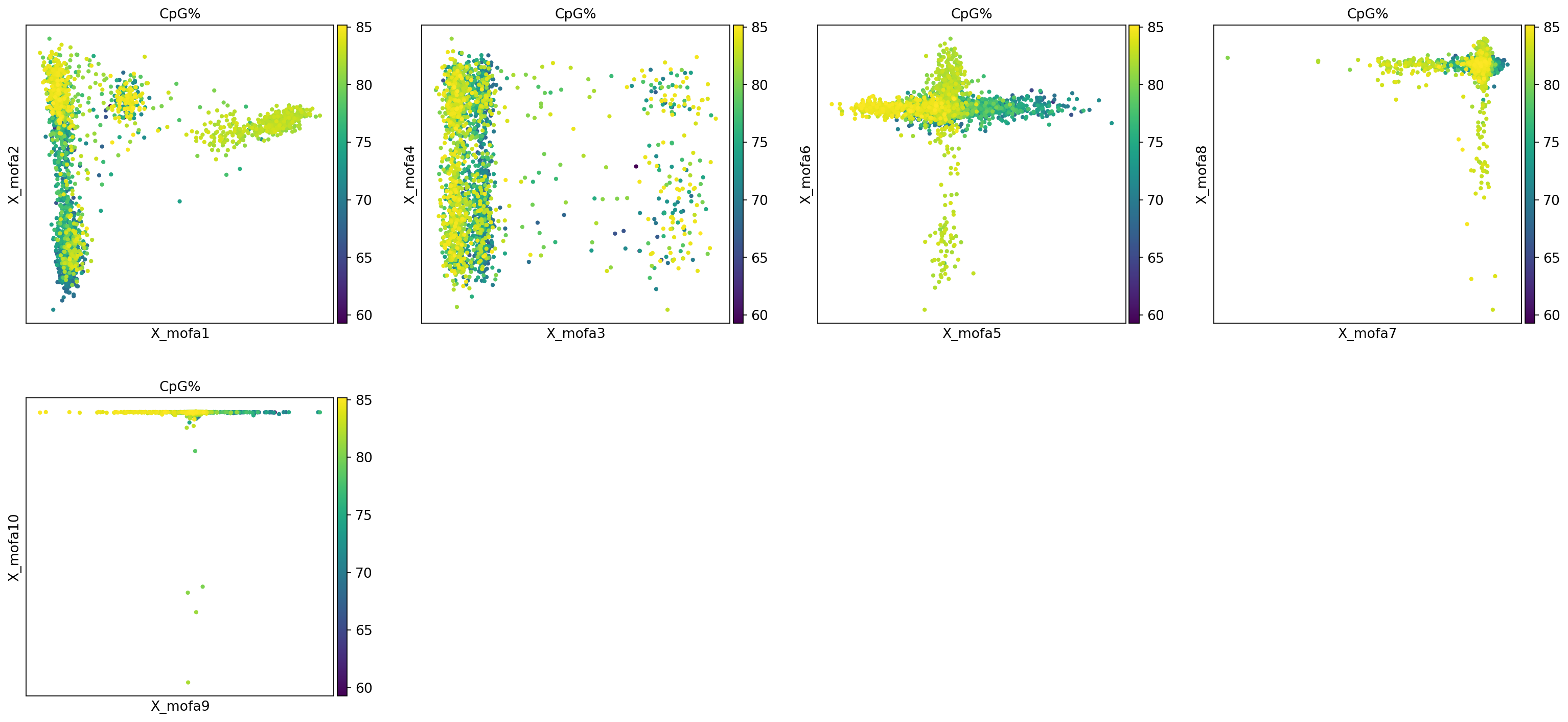
Model Evaluation and Variance Explanation (R2)
Assess the explanation degree (Variance Explained, R2) of each factor for the variation of different omics modalities.
model = mfx.mofa_model(f"{outdir}/models/pbmc_969_multi_methy.hdf5")
modelSamples (cells): 2189
Features: 4500
Groups: sample_WTJW969_E (2189)
Views: methy_geneslop2k (2500), rna (2000)
Factors: 10
Expectations: W, Z
model.get_r2()| Factor | View | Group | R2 | |
|---|---|---|---|---|
| 0 | Factor1 | rna | sample_WTJW969_E | 1.755070e+01 |
| 1 | Factor1 | methy_geneslop2k | sample_WTJW969_E | 1.526547e-06 |
| 2 | Factor2 | rna | sample_WTJW969_E | 6.230776e+00 |
| 3 | Factor2 | methy_geneslop2k | sample_WTJW969_E | 1.247076e-05 |
| 4 | Factor3 | rna | sample_WTJW969_E | 5.786080e+00 |
| 5 | Factor3 | methy_geneslop2k | sample_WTJW969_E | 6.985795e-07 |
| 6 | Factor4 | rna | sample_WTJW969_E | 5.757473e-06 |
| 7 | Factor4 | methy_geneslop2k | sample_WTJW969_E | 2.565718e+00 |
| 8 | Factor5 | rna | sample_WTJW969_E | 1.429741e+00 |
| 9 | Factor5 | methy_geneslop2k | sample_WTJW969_E | 1.103752e-04 |
| 10 | Factor6 | rna | sample_WTJW969_E | 9.622432e-01 |
| 11 | Factor6 | methy_geneslop2k | sample_WTJW969_E | 7.399176e-06 |
| 12 | Factor7 | rna | sample_WTJW969_E | 8.270760e-01 |
| 13 | Factor7 | methy_geneslop2k | sample_WTJW969_E | 2.246223e-06 |
| 14 | Factor8 | rna | sample_WTJW969_E | 5.685163e-01 |
| 15 | Factor8 | methy_geneslop2k | sample_WTJW969_E | 3.142382e-06 |
| 16 | Factor9 | rna | sample_WTJW969_E | 4.485491e-01 |
| 17 | Factor9 | methy_geneslop2k | sample_WTJW969_E | 2.246235e-05 |
| 18 | Factor10 | rna | sample_WTJW969_E | 8.303199e-02 |
| 19 | Factor10 | methy_geneslop2k | sample_WTJW969_E | -2.510329e-07 |
r2_df = model.get_r2()
r2_df[r2_df['R2'] >= 1]| Factor | View | Group | R2 | |
|---|---|---|---|---|
| 0 | Factor1 | rna | sample_WTJW969_E | 17.550695 |
| 2 | Factor2 | rna | sample_WTJW969_E | 6.230776 |
| 4 | Factor3 | rna | sample_WTJW969_E | 5.786080 |
| 7 | Factor4 | methy_geneslop2k | sample_WTJW969_E | 2.565718 |
| 8 | Factor5 | rna | sample_WTJW969_E | 1.429741 |
Figure Legend: This heatmap shows the explanatory power of each MOFA factor (Factor) for the variation of different omics modalities (View).
- X-axis: MOFA Factors (Factor 1, 2, ...).
- Y-axis: Omics Modalities (RNA, Methylation).
- Color Intensity: R2 value (Variance Explained), darker color indicates greater contribution of the factor to that modality.
- Significance: If a factor is dark in both modalities, it is usually a Shared Factor; if dark in only one modality, it is usually a Private Factor.
mfx.plot_r2(model, x='View', vmax=10)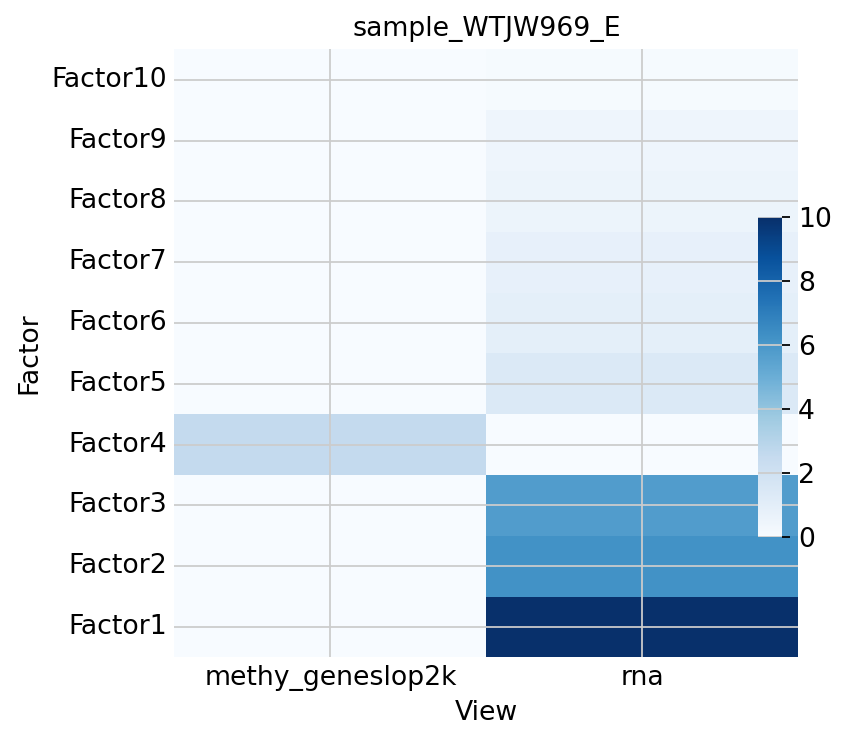
model.close()Downstream Analysis Based on MOFA Factors
Use MOFA extracted latent factors (X_mofa) to replace original features, construct a neighbor graph, and perform UMAP dimensionality reduction and Leiden clustering. Combine with transcriptome annotation results to assess if a clearer cell atlas can be obtained.
In this step, we typically select factors with high variance explanation for subsequent analysis.
sc.pp.neighbors(mdata, use_rep="X_mofa", n_pcs=5, random_state=20)
sc.tl.umap(mdata, min_dist=.2, spread=1., random_state=20)
sc.tl.leiden(mdata, resolution=.5, random_state=20)Figure Legend: This figure shows the UMAP dimensionality reduction plot constructed based on MOFA factors.
- This figure integrates information from RNA and Methylation, usually distinguishing cell subpopulations better than single-modality clustering.
- Color Legend:
celltype: Cell type annotation based on transcriptome data.leiden: Clustering results based on MOFA factors.CpG%: Genome-wide CpG methylation level.Sample: Sample source.
sc.pl.umap(mdata, color=["celltype","leiden",'CpG%', 'Sample'],legend_loc="on data", legend_fontoutline=1)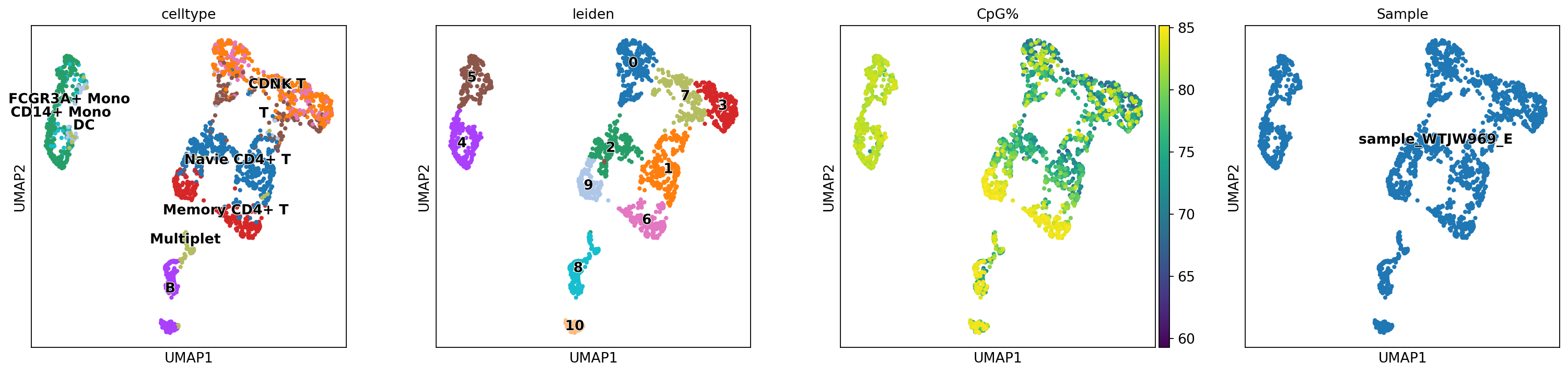
Results Discussion and Optimization Suggestions
MOFA+ performs subsequent multi-modal integration analysis based on highly variable features, so it is significantly affected by the selected High Variable Feature Set. If the selected high variable feature set itself cannot correctly distinguish different biological cell groups, the integration result of MOFA+ will also be limited.
As shown in the figure above, if the multi-modal integration clustering effect of MOFA+ is not as good as the single-omics (e.g., RNA only) clustering result, a possible reason is that the methylation region granularity we used is "too large" (e.g., geneslop2k refers to the gene region plus its upstream and downstream 2kb). Existing literature indicates that methylation levels in different gene regions (such as promoters, enhancers, gene bodies) have different regulatory meanings. Coarse-grained regions may mask fine biological signals, leading to the selected high variable feature set containing more noise, thereby affecting the integration effect of MOFA+.
Optimization Strategy: Successful cases of integrating methylation and transcriptome data using MOFA+ indicate that precise screening of methylation feature sets is crucial. For example, in the literature Multi-omics profiling of mouse gastrulation at single-cell resolution, researchers adopted the following strategy:
- Feature Pre-screening: Use public data or ChIP-seq data to first locate key regulatory regions related to sample types, such as
H3K27ac(Enhancers),H3K4me3(TSS), and 2kb regions upstream and downstream of protein-coding gene transcription start sites (Promoters). - High Variable Screening: Perform high variable feature screening based on these selected regions.
- Factor Discovery: Identify factors strongly related to biological processes in the methylation modality.
By this method, the signal-to-noise ratio of input features can be significantly improved, thereby enhancing the effect of MOFA+ integration analysis.
Output Files and Subsequent Reuse
After analysis is complete, we write three objects to outdir:
adata_rna.h5ad: RNAAnnDataobject.adata_met_**.h5ad: MethylationAnnDataobject.mdata.h5mu: Multi-omicsMuDataobject.
Note:
.h5mufiles are usually large, it is recommended to keep them in a stable path and back them up.- If you change filtering thresholds or
var_dimparameters, it is recommended to rerun the full workflow and overwrite output files.
adata_rna.write_h5ad(f'{outdir}/adata_rna.h5ad')
for i in adata_met_list.keys():
adata_met_list[i].write_h5ad(f'{outdir}/adata_met_{i}.h5ad')
mdata.write(f'{outdir}/mdata.h5mu')More Resources
- Muon Official Tutorial: For other analysis tutorials on integrating multi-modal data with MOFA+, please see the Muon Website.
- MOFA+ Downstream Analysis: For downstream analysis after MOFA+ (such as factor annotation, gene set enrichment, etc.), please see the mofax tutorial.