甲基化 + RNA 双组学: 基于 MOFA+ 算法的单细胞甲基化与转录组多组学整合分析教程
模块简介
本教程详细介绍了如何基于 MOFA+ (Multi-Omics Factor Analysis v2) 框架,对单细胞甲基化 (DNA Methylation) 和转录组 (RNA) 数据进行多组学整合分析。
MOFA+ 分析原理
MOFA+ 是一种无监督的统计框架,旨在从多组学数据中推断潜在的低维因子 (Latent Factors)。其核心思想是将不同组学模态 (Views) 的数据分解为一组共享或私有的潜在因子。
- 输入数据 (Input Data):包含多个组学模态的矩阵(如 RNA 和 DNA Methylation),矩阵的行代表对齐的细胞,列代表各自的特征(基因或甲基化区域)。
- 潜在因子 (Latent Factors):模型通过矩阵分解提取出驱动数据变异的低维因子。
- 共享因子 (Shared Factors):捕获在多个组学模态中共同存在的生物学变异(如细胞类型、细胞周期等),这些因子同时解释 RNA 和甲基化数据的方差。
- 私有因子 (Private Factors):捕获仅存在于单一组学模态中的变异(如特定组学的技术噪声、批次效应或特异性生物学信号)。
- 模型重构 (Model Reconstruction):数据被分解为因子矩阵 (Factor Matrix) 和权重矩阵 (Weight Matrix) 的乘积。权重矩阵衡量了每个特征对特定因子的贡献度。
- 稀疏性 (Sparsity):MOFA+ 引入了稀疏性先验,使得每个因子仅与少量的关键特征(基因或甲基化区域)相关联。这种正则化不仅防止了过拟合,还显著提高了模型的生物学可解释性。
分析目标
- 多组学降维:将高维的 RNA 和甲基化数据压缩成少量的潜在因子,保留主要的生物学变异。
- 变异解释:量化并评估每个因子对不同组学模态整体方差的贡献度 (Variance Explained)。
- 联合聚类:基于提取的低维因子空间构建邻居图,进行更稳健的联合降维 (UMAP) 和细胞分群 (Leiden)。
准备工作
导入依赖库与定义辅助函数
本部分代码主要完成以下任务:
- 导入分析库:包括单细胞分析标准库 (
scanpy,muon)、甲基化分析专用库 (ALLCools) 以及绘图库 (matplotlib)。 - 定义数据加载与处理函数:
mcds_to_adata: 将 MCDS 格式(甲基化存储格式)转换为AnnData对象。mcds_get_mc_frac_adata: 读取并处理甲基化率(Methylation Fraction)矩阵,支持黑名单区域过滤和高变特征计算。compute_cg_ch_level: 计算每个细胞的全局 CG 和 CH 甲基化水平。check_barcode_overlap: 检查 RNA 和 Methylation 数据的细胞 Barcode 重叠情况,确保多组学数据来自同一批细胞。parse_gtf: 解析 GTF 注释文件,提取基因类型信息。
import os
# 设置环境变量,确保调用正确的 conda 环境和二进制文件
os.environ['PATH'] = "/PROJ/home/weiqiuxia/micromamba/envs/muon_env/bin:/opt/conda/bin:/opt/conda/condabin:/usr/local/sbin:/usr/local/bin:/usr/sbin:/usr/bin:/sbin:/bin:/PROJ2/FLOAT/public/bin"
r_base_path = '/PROJ/home/weiqiuxia/micromamba/envs/muon_env'
os.environ['R_HOME'] = os.path.join(r_base_path, 'lib', 'R')
# 设置动态库路径
os.environ['LD_LIBRARY_PATH'] = os.path.join(r_base_path, 'lib', 'R', 'lib') + ':' + os.environ.get('LD_LIBRARY_PATH', '')
# 设置R包库路径
r_lib_path = os.path.join(r_base_path, 'lib', 'R', 'library')
os.environ['R_LIBS'] = r_lib_path
os.environ['R_LIBS_USER'] = r_lib_path
os.environ['R_LIBS_SITE'] = r_lib_path
import re
import glob
from ALLCools.mcds import MCDS
from ALLCools.clustering import tsne, significant_pc_test, log_scale, lsi, binarize_matrix, filter_regions, cluster_enriched_features, ConsensusClustering, Dendrogram, get_pc_centers
from ALLCools.clustering.doublets import MethylScrublet
from ALLCools.plot import *
import scanpy as sc
import scanpy.external as sce
from harmonypy import run_harmony
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
from matplotlib.lines import Line2D
import warnings
import xarray as xr
from ALLCools.clustering import one_vs_rest_dmg
from scipy import sparse
import muon as mu
from muon import atac as ac
from skmisc.loess import loess
import mofax as mfx
import re
from collections import defaultdict
sc.settings.verbosity = 0
warnings.filterwarnings('ignore')
try:
from matplotlib_venn import venn2
HAS_VENN = True
except Exception:
HAS_VENN = False
plt.rcParams['figure.dpi'] = 80
plt.rcParams['savefig.dpi'] = 300
plt.rcParams['figure.figsize'] = (5,5)from numcodecs.checksum32 import CRC32, Adler32, JenkinsLookup3
/PROJ/home/weiqiuxia/micromamba/envs/muon_env/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
/PROJ/home/weiqiuxia/micromamba/envs/muon_env/lib/python3.12/site-packages/muon/_core/preproc.py:31: FutureWarning: \`__version__\` is deprecated, use \`importlib.metadata.version('scanpy')\` instead
if Version(scanpy.__version__) < Version("1.10"):
def mcds_to_adata(mcds_path, var_dim = 'chrom20k', mc_type = 'CGN', quant_type = 'hypo-score'):
'''
将 hypo-score 矩阵转换为 AnnData 对象
'''
# 读取 MCDS 文件
mcds = MCDS.open(mcds_path, obs_dim = 'cell', var_dim = var_dim)
# 获取评分矩阵并转换为 AnnData
adata = mcds.get_score_adata(mc_type=mc_type, quant_type=quant_type, sparse = True)
return(adata)
def mcds_get_mc_frac_adata(mcds_path, var_dim = 'chrom20k', mc_type = 'CGN', obs_dim = 'cell',
min_cov = 1, met_matrix_type = 'mc_frac', hvf_method='SVR' ,n_top_feature = 2000,
clip_norm_value = 10,
black_list_path = None, ):
'''
将甲基化率矩阵或 M-value 转换为 AnnData 对象
'''
# 1. 打开 MCDS 数据集
mcds = MCDS.open(
mcds_path,
obs_dim = 'cell',
var_dim = var_dim
)
# 2. 计算特征的平均覆盖度 (Coverage Mean)
if "mc_type" in mcds[f'{var_dim}_da'].dims:
feature_cov_mean = mcds.sel(count_type="cov").sum(dim="mc_type").mean(dim=obs_dim).squeeze().to_pandas()
else:
feature_cov_mean = mcds[f'{var_dim}_da'].sel(count_type="cov").mean(dim=obs_dim).squeeze().to_pandas()
mcds.coords[f'{var_dim}_cov_mean'] = (var_dim, feature_cov_mean)
# 过滤掉低覆盖度的特征
mcds = mcds.filter_feature_by_cov_mean(min_cov=min_cov)
# 3. (可选) 移除黑名单区域 (Blacklist Regions)
if black_list_path:
mcds = mcds.remove_black_list_region(
black_list_path = black_list_path,
f = 0.2
)
load = True
obs_dim = 'cell'
# 4. 计算甲基化矩阵 (M-value 或 Methylation Fraction)
if met_matrix_type == 'mvalue':
mcds.add_m_value(var_dim=var_dim)
da_suffix = 'mvalue'
else:
if met_matrix_type == 'mc_frac':
mcds.add_mc_frac(var_dim=var_dim,clip_norm_value=clip_norm_value,normalize_per_cell=True)
else:
clip_norm_value = None
mcds.add_mc_frac(var_dim=var_dim,clip_norm_value=clip_norm_value,normalize_per_cell=False)
da_suffix = 'frac'
# 加载数据到内存
if load and (mcds.get_index(obs_dim).size < 20000):
mcds[f'{var_dim}_da_{da_suffix}'].load()
# 5. 计算高变特征 (Highly Variable Features)
if hvf_method == 'SVR':
mcg_hvf = mcds.calculate_hvf_svr(
var_dim = var_dim,
mc_type = mc_type,
da_suffix = da_suffix,
n_top_feature = n_top_feature,
plot = False)
else:
mcg_hvf = mcds.calculate_hvf(
var_dim = var_dim,
mc_type = mc_type,
da_suffix = da_suffix,
n_top_feature = n_top_feature,
plot = False)
# 6. 生成 AnnData 对象并合并高变特征信息
mcg_adata = mcds.get_adata(
mc_type=mc_type,
var_dim = var_dim,
select_hvf = True,
da_suffix = da_suffix)
mcg_adata.var = mcg_adata.var.merge(mcg_hvf, right_index = True, left_index = True)
return mcg_adata
def compute_cg_ch_level(df):
'''
计算 CG 和 CH 甲基化水平
'''
# 筛选 CG 和 CH 上下文的覆盖度 (cov) 和甲基化 (mc) 列
cg_cov_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_cov')]
cg_mc_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_mc')]
ch_cov_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_cov')]
ch_mc_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_mc')]
# 计算全局 CG 甲基化率
df['CpG_cov'] = df[cg_cov_col].sum(axis=1)
df['CpG_mc'] = df[cg_mc_col].sum(axis=1)
df['CpG%'] = round(df['CpG_mc'] * 100 / df['CpG_cov'], 2)
# 计算全局 CH 甲基化率
df['CH_cov'] = df[ch_cov_col].sum(axis=1)
df['CH_mc'] = df[ch_mc_col].sum(axis=1)
df['CH%'] = round(df['CH_mc'] * 100 / df['CH_cov'], 2)
return df
def check_barcode_overlap(rna_adata, met_adata, barcode_map = None):
'''
检查转录组和甲基化 Barcode 的重叠情况,并将转录组 Barcode 重置为对应的甲基化 Barcode。
'''
# 计算 Barcode 重叠率
rna_adata.obs['original_barcode'] = [ re.sub('_.*', '', i) for i in rna_adata.obs.index ]
met_adata.obs['original_barcode'] = [ re.sub('_.*', '', i) for i in met_adata.obs.index ]
overlap_rate = len(set(rna_adata.obs['original_barcode']) & set(met_adata.obs['original_barcode'])) / met_adata.shape[0]
# 如果重叠率低 (<10%),尝试使用映射表转换 Barcode
if overlap_rate < 0.1:
met2rna_barcode = barcode_map[barcode_map['m_cb'].isin(met_adata.obs['original_barcode'])]
rna_adata = rna_adata[rna_adata.obs['original_barcode'].obs.index.isin(met2rna_barcode['gex_cb'])]
met2rna_barcode = met2rna_barcode.set_index('gex_cb')
rna_adata.obs['gex_cb'] = rna_adata.obs.index
rna_adata.obs.index = met2rna_barcode.loc[rna_adata.obs['original_barcode'],'m_cb']
# 再次检查重叠率
overlap_rate_test = len(set(rna_adata.obs.index) & set(met_adata.obs.index)) / met_adata.shape[0]
chemistry = 'DD-MET3'
if overlap_rate_test < 0.1:
raise Exception('The overlap rate between RNA data and MET data barcode is less than 10%')
else:
chemistry = 'DD-MET5'
rna_adata.obs.index = rna_adata.obs['original_barcode']
met_adata.obs.index = met_adata.obs['original_barcode']
# 仅保留两个模态共有的细胞
common_index = list(set(rna_adata.obs.index) & set(met_adata.obs.index))
rna_adata = rna_adata[rna_adata.obs.index.isin(common_index)]
rna_adata.obs['chemistry'] = chemistry
met_adata = met_adata[met_adata.obs.index.isin(common_index)]
met_adata.obs['chemistry'] = chemistry
return rna_adata, met_adata, chemistry
def parse_gtf(gtf_file, output_file):
'''
Read GTF, get gene_type information for genes.
'''
with open(gtf_file, 'r') as f_in, open(output_file, 'w') as f_out:
for line in f_in:
if line.startswith('#'):
continue
parts = line.strip().split('\t')
if len(parts) < 9:
continue
feature_type = parts[2]
if feature_type != 'gene':
continue
attributes = parts[8]
attr_dict = {}
# Basic parsing of GTF attributes: key "value"; key "value";
for attr in attributes.split(';'):
attr = attr.strip()
if not attr:
continue
if ' ' in attr:
key, value = attr.split(' ', 1)
value = value.strip('"')
attr_dict[key] = value
gene_id = attr_dict.get('gene_id', '')
gene_name = attr_dict.get('gene_name', '')
gene_type = attr_dict.get('gene_type', '')
# Construct the first column
if gene_id and gene_name:
col1 = f"{gene_id}_{gene_name}"
elif gene_id:
col1 = gene_id
else:
col1 = gene_name # Should not happen based on GTF spec but good to handle
f_out.write(f"{col1}\t{gene_type}\n")
def rds_to_matrix(filepath, outdir):
import rpy2.robjects as robjects
robjects.globalenv['rdsfile'] = filepath
robjects.globalenv['outdir'] = outdir
print(f"R_HOME: {os.environ['R_HOME']}")
print(f"R_LIBS: {os.environ['R_LIBS']}")
print(f"LD_LIBRARY_PATH: {os.environ['LD_LIBRARY_PATH']}")
# 执行R代码并获取结果
robjects.r('''
library(Seurat)
library(qs)
if(endsWith(rdsfile, ".rds")){
obj <- readRDS(rdsfile)
}else{
obj <- qread(rdsfile)
}
dir.create(outdir, recursive = TRUE)
Matrix::writeMM(GetAssayData(obj, assay = "RNA", layer = "counts"), file = file.path(outdir, "matrix.mtx") )
cmd <- paste0("gzip ", file.path(outdir, "matrix.mtx"))
system(cmd)
features <- data.frame(Feature_id = rownames(obj), Feature_name = rownames(obj), Feature_type = "Gene Expression")
write.table(features, file.path(outdir, "features.tsv"), row.names = F, col.names = F, sep = "\t", quote = F)
cmd <- paste0("gzip ", file.path(outdir, "features.tsv"))
system(cmd)
barcodes <- data.frame(Barcode = colnames(obj))
write.table(barcodes, file.path(outdir, "barcodes.tsv"), row.names = F, col.names = F, sep = "\t", quote = F)
cmd <- paste0("gzip ", file.path(outdir, "barcodes.tsv"))
system(cmd)
meta <- obj@meta.data
write.table(tibble::rownames_to_column(meta, "barcode"), file.path(outdir, "meta.csv"), row.names = FALSE, col.names = TRUE, sep = ",", quote = F)
''')输入目录结构说明
重要提示:本流程对输入数据的目录结构有严格要求。程序将根据 indir 参数自动检索 RNA 和甲基化相关的输入文件。建议按照以下结构组织数据:
data/AY1768874914782/ (indir)
├── input.rds # 转录组数据(Seurat 对象或 qs 格式)
└── methylation # 甲基化数据根目录
└── demoWTJW969-task-1 # 任务/批次名称
└── WTJW969 # 样本名称
├── allcools_generate_datasets
│ └── WTJW969.mcds # MCDS 数据集文件夹
│ ├── chrom100k
│ ├── chrom10k
│ ├── chrom20k # 20kb 窗口的甲基化得分矩阵
│ └── ...
└── split_bams
├── filtered_barcode_reads_counts.csv # 过滤后的 Barcode mapped reads 计数
└── WTJW969_cells.csv # 过滤后的细胞质控信息(CpG 数量、覆盖度等)- RNA 数据检索:在
indir路径下匹配.rds或.qs文件。 - 甲基化数据检索:在
indir/methylation路径下检索 MCDS 文件及关联的质控 CSV 文件。
# indir:输入目录
indir = '/home/weiqiuxia/workspace/data/AY1768874914782/'
# outdir:结果输出目录
outdir = '/home/weiqiuxia/workspace/project/weiqiuxia/MOFA_results'
# dd_met3_barcode_map:DD-MET3 的 barcode 映射表
dd_met3_barcode_map = '/home/weiqiuxia/workspace/project/weiqiuxia/DD-M_bUCB3_whitelist.csv'
# black_list_path:需要过滤的区域,bed格式文件。甲基化数据中,会将与以下设置的bed区域有20%overlap的区域过滤掉
black_list_path = '/home/weiqiuxia/workspace/project/weiqiuxia/hg38-blacklist-sort.v2.bed'
# resolution:甲基化聚类的分辨率 (resolution),浮点数,需大于 0。
resolution = 1
# anno_col:RNA 元数据中的细胞注释列名
anno_col = 'CellAnnotation'
# sample_col:样本列名,默认 `Sample`;需与 `indir` 路径中的样本名一致。
sample_col = 'Sample'
# rna_n_top_feature:转录组的高变基因数量
rna_n_top_feature = 2000
# matrix_type:甲基化使用的 matrix参数。可选的值有:hypo-score|mvalue
met_matrix_type = 'mvalue'
# var_dim:甲基化特征维度。可选多个
var_dim = ['geneslop2k']
# total_cpg_number_cutoff:CpG 数量阈值
total_cpg_number_cutoff = 500
# mc_type:甲基化的类型:CGN。目前只支持 CGN
mc_type = 'CGN'
# min_cov:过滤掉平均coverage小于此值的features。
min_cov = 10
# normalize_per_cell:是否甲基化率的后验估计。True对原始的甲基化水平进行去噪和插补,False则使用原始的甲基化水平
normalize_per_cell=True
# met_n_top_feature:筛选高变feature的方法。可能的值有:SVR|Based
hvf_method='SVR'
# met_n_top_feature:甲基化数据需要筛选的高变features的数量
met_n_top_feature = 2500
# total_cpg_number_cutoff:CpG 数量阈值
total_cpg_number_cutoff = 500os.makedirs(outdir, exist_ok=True)# 读取 DD-MET3 barcode 映射表
dd_met3_barcode_map_df = pd.read_csv(dd_met3_barcode_map, header = 0)# 首先获取qs或者rds结尾的文件,并转成矩阵文件,然后使用scanpy读取
for i in os.listdir(indir):
if i.endswith('.qs'):
rds_to_matrix(os.path.join(indir, i), os.path.join(outdir, 'input_matrix'))
breakR callback write-console: Loading required package: sp
R_HOME: /PROJ/home/weiqiuxia/micromamba/envs/muon_env/lib/R
R_LIBS: /PROJ/home/weiqiuxia/micromamba/envs/muon_env/lib/R/library
LD_LIBRARY_PATH: /PROJ/home/weiqiuxia/micromamba/envs/muon_env/lib/R/lib:
R callback write-console:
Attaching package: ‘SeuratObject’
R callback write-console: The following objects are masked from ‘package:base’:
intersect, t
R callback write-console: qs 0.27.3. Announcement: https://github.com/qsbase/qs/issues/103
# scanpy 读取 RNA 数据
adata_rna = sc.read_10x_mtx(f'{outdir}/input_matrix/')
meta = pd.read_csv(f'{outdir}/input_matrix/meta.csv', index_col = 0, header = 0)
adata_rna.obs = meta
adata_rna.obs['CellAnnotation'] = adata_rna.obs['CellAnnotation'].astype('category')甲基化数据预处理
单细胞甲基化数据具有高度稀疏性和不同的信号分布特征,因此需要针对指定的甲基化矩阵类型 (met_matrix_type) 采取特定的预处理策略,以确保下游分析的准确性。
我们支持以下两种常见的数据类型及其对应的处理流程:
Hypo-score 矩阵:
- 二值化 (Binarization):使用
binarize_matrix函数将连续的 Hypo-score 转换为二值信号。我们将hypo-score > 0.95的位点视为显著低甲基化(标记为 1),其余视为背景(标记为 0)。这有助于降低技术噪声并突显生物学信号。 - TF-IDF 转换:应用 TF-IDF (Term Frequency-Inverse Document Frequency) 算法。这是处理稀疏二值数据的标准方法(类似于 scATAC-seq 分析),能有效校正测序深度差异,并降低普遍存在特征的权重,从而提取细胞特异性信息。
- 二值化 (Binarization):使用
M-value 矩阵:
- Logit 变换:基于原始甲基化率 (Fraction),通过公式
np.log2((frac + alpha) / (1 - frac + alpha))计算 M-value。这种变换可以将区间 [0, 1] 的甲基化率映射到实数域,且具有更好的方差稳定性,更适合用于线性模型和降维分析。
- Logit 变换:基于原始甲基化率 (Fraction),通过公式
随后,我们将基于处理后的矩阵进行高变特征筛选 (Feature Selection),即筛选出在细胞间变异最大的甲基化区域,为后续的多模态整合做好准备。
#读取甲基化数据,然后与转录组数据取交集,确保甲基化和转录组的数据的细胞一致,barcode一致
keep_col = ['barcode', 'total_cpg_number', 'genome_cov', 'reads_counts', 'CpG%', 'CH%']
obj_met_list = defaultdict(defaultdict)
obj_rna_list = defaultdict()
indirs = indir.split(',')
samples = []
mcds_paths = {}
sample_path = {}
analysis_batch_dir = os.listdir(os.path.join(indir, 'methylation'))
for i in analysis_batch_dir:
if os.path.isdir(os.path.join(indir, 'methylation', i)):
a_batch_dir = os.path.join(indir, 'methylation', i)
# 获取分析批次下所有样本
samples.extend([ i for i in os.listdir(a_batch_dir)])
for s in samples:
mcds_path = os.path.join(a_batch_dir, f'{s}','allcools_generate_datasets', f'{s}.mcds')
mcds_paths[s] = mcds_path
sample_path[s] = a_batch_dir
# 读取甲基化相关的质控结果
s_cells_meta = pd.read_csv(os.path.join(a_batch_dir, s, 'split_bams', f'{s}_cells.csv'), header = 0)
s_cells_meta['barcode'] = [b.replace('_allc.gz','') for b in s_cells_meta['cell_barcode']]
# 读取每个细胞的mapped reads
s_reads_counts = pd.read_csv(os.path.join(a_batch_dir, s, 'split_bams', 'filtered_barcode_reads_counts.csv'), header = 0)
s_merged_df = s_cells_meta.merge(s_reads_counts, how = 'inner', left_on = 'barcode', right_on = 'barcode')
# 合并以上两个dataframe
s_merged_df = compute_cg_ch_level(s_merged_df)
s_merged_df = s_merged_df[keep_col]
s_merged_df = s_merged_df.set_index('barcode')
#读取 Methylation 矩阵并构建 AnnData
if met_matrix_type == 'hypo-score':
# 处理 hypo-score 类型数据
if isinstance(var_dim, list):
for vd in var_dim:
# 将 MCDS 转换为 AnnData
obj = mcds_to_adata(
mcds_path,
var_dim = vd)
# 合并元数据
obj.obs = obj.obs.merge(s_merged_df, left_index = True, right_index = True)
# 过滤低质量细胞
obj = obj[obj.obs['total_cpg_number'] >= total_cpg_number_cutoff ]
obj.obs['Sample'] = s
# 检查同一个样本的转录组数据和甲基化数据的细胞barcode是否一致,细胞数量是否一致;取交集
adata_rna_s = adata_rna[adata_rna.obs['orig.ident'] == s]
obj_rna_list[s], obj, chemistry = check_barcode_overlap(adata_rna_s, obj, dd_met3_barcode_map_df)
obj_met_list[vd][s] = obj
else:
obj = mcds_to_adata(
mcds_path,
var_dim = var_dim)
obj.obs = obj.obs.merge(s_merged_df, left_index = True, right_index = True)
obj = obj[obj.obs['total_cpg_number'] >= total_cpg_number_cutoff ]
obj.obs['Sample'] = s
adata_rna_s = adata_rna[adata_rna.obs['orig.ident'] == s]
obj_rna_list[s], obj, chemistry = check_barcode_overlap(adata_rna_s, obj, dd_met3_barcode_map_df)
obj_met_list[var_dim][s] = obj
else:
# 处理 mc_frac 或 mvalue 类型数据
if isinstance(var_dim, list):
for vd in var_dim:
obj = mcds_get_mc_frac_adata(
mcds_path,
var_dim = vd,
mc_type = mc_type,
min_cov = min_cov,
met_matrix_type = met_matrix_type,
hvf_method=hvf_method ,
n_top_feature = met_n_top_feature,
black_list_path = black_list_path
)
obj.obs = obj.obs.merge(s_merged_df, left_index = True, right_index = True)
obj = obj[obj.obs['total_cpg_number'] >= total_cpg_number_cutoff ]
obj.obs['Sample'] = s
adata_rna_s = adata_rna[adata_rna.obs['orig.ident'] == s]
obj_rna_list[s], obj, chemistry = check_barcode_overlap(adata_rna_s, obj, dd_met3_barcode_map_df)
obj_met_list[vd][s] = obj
else:
obj = mcds_get_mc_frac_adata(
mcds_path,
var_dim = var_dim,
mc_type = mc_type,
min_cov = min_cov,
met_matrix_type = met_matrix_type,
hvf_method=hvf_method ,
n_top_feature = met_n_top_feature,
black_list_path = black_list_path
)
obj.obs = obj.obs.merge(s_merged_df, left_index = True, right_index = True)
obj = obj[obj.obs['total_cpg_number'] >= total_cpg_number_cutoff ]
obj.obs['Sample'] = s
adata_rna_s = adata_rna[adata_rna.obs['orig.ident'] == s]
obj_rna_list[s], obj, chemistry = check_barcode_overlap(adata_rna_s, obj, dd_met3_barcode_map_df)
obj_met_list[var_dim][s] = obj
else:
raise Exception('file not found!')After cov mean filter: 25666 geneslop2k 66.5%
261 geneslop2k features removed due to overlapping (bedtools intersect -f 0.2) with black list regions.
Fitting SVR with gamma 0.0417, predicting feature dispersion using mc_frac_mean and cov_mean.
Total Feature Number: 25405
Highly Variable Feature: 2500 (9.8%)
select_hvf is True, but no highly variable feature results found, use all features instead.
# 如果是有多个样本,需要将矩阵合并
if len(obj_rna_list) > 1:
adata_rna = obj_rna_list[samples[0]].concatenate([obj_rna_list[i] for i in samples[1:] ])
else:
adata_rna = obj_rna_list[samples[0]]
del obj_rna_list
adata_met_list = {}
for i in obj_met_list.keys():
if len(obj_met_list[i]) > 1:
adata_met_list[i] = obj_met_list[i][samples[0]].concatenate([obj_met_list[i][s] for s in samples[1:] ])
else:
adata_met_list[i] = obj_met_list[i][samples[0]].copy()
del obj_met_listfor i in adata_met_list.keys():
if met_matrix_type == 'hypo-score':
# 1. 二值化处理:将连续的 hypo-score 转换为二值信号 (0/1),cutoff=0.95
binarize_matrix(adata_met_list[i], cutoff=0.95)
# 2. 区域过滤:过滤掉低质量或无信息的基因组区域
filter_regions(adata_met_list[i])
# 保存原始数据到 layers
adata_met_list[i].layers['raw'] = adata_met_list[i].X.copy()
# 3. TF-IDF 归一化:校正测序深度,突出特异性信号 (scale_factor=1e4)
ac.pp.tfidf(adata_met_list[i], scale_factor=1e4)
# 4. 特征选择:选择高变异的甲基化区域 (Highly Variable Features)
sc.pp.highly_variable_genes(adata_met_list[i], flavor='seurat_v3', n_top_genes=met_n_top_feature, batch_key='Sample')
# 5. LSI 降维:基于 SVD 的降维方法,适用于稀疏数据
ac.tl.lsi(adata_met_list[i])
else:
# 非 hypo-score 数据的处理逻辑
min_samples = 2 if adata_met_list[i].obs[sample_col].value_counts().shape[0] > 1 else 1
feature_select_cols = [col for col in adata_met_list[i].var.columns if col.startswith('feature_select')]
adata_met_list[i].var['highly_variable'] = adata_met_list[i].var[feature_select_cols].sum(axis=1) >= min_samplesRNA 数据预处理
RNA 数据的预处理遵循标准的 Scanpy 流程,旨在去除噪音并保留生物学信号:
- 质量控制 (QC):
sc.pp.filter_cells:过滤掉检测到的基因数过少的低质量细胞。sc.pp.filter_genes:过滤掉在极少数细胞中表达的基因。
- 归一化 (Normalization):
sc.pp.normalize_total:消除由测序深度(库大小)差异带来的技术影响,通常将每个细胞标准化到 10,000 reads。
- 对数变换 (Log Transformation):
sc.pp.log1p:进行变换。这能使数据分布更接近正态分布,稳定方差,并压缩表达量的动态范围。
- 高变基因选择 (Feature Selection):
sc.pp.highly_variable_genes:筛选出生物学变异显著的高变基因 (HVGs)。这些基因包含了区分不同细胞类型的主要信息,用于后续的降维分析。
# 标记线粒体基因 (Mitochondrial genes)
# 人类使用 "MT-",小鼠使用 "Mt-"
adata_rna.var["mt"] = adata_rna.var_names.str.startswith("MT-")
# 标记核糖体基因 (Ribosomal genes)
adata_rna.var["ribo"] = adata_rna.var_names.str.startswith(("RPS", "RPL"))
# 标记血红蛋白基因 (Hemoglobin genes)
adata_rna.var["hb"] = adata_rna.var_names.str.contains("^HB[^(P)]")
# 计算 QC 指标 (Quality Control Metrics)
# qc_vars: 需要计算百分比的基因集合 (如线粒体比例)
sc.pp.calculate_qc_metrics(adata_rna, qc_vars=["mt", "ribo", "hb"], inplace=True, log1p=True)# 1. 备份原始计数数据
adata_rna.layers["counts"] = adata_rna.X.copy()
# 2. 总数归一化 (Normalization)
# 将每个细胞的文库大小标准化为中位数,消除测序深度差异
sc.pp.normalize_total(adata_rna)
# 3. 对数变换 (Log transformation)
# 对数据取 log(x+1),使数据分布更接近正态分布,缓解偏度
sc.pp.log1p(adata_rna)
# 4. 筛选高变基因 (Highly Variable Genes)
# 识别在细胞间变异最大的基因,用于后续降维和聚类
sc.pp.highly_variable_genes(adata_rna, flavor='seurat_v3', n_top_genes=2000, batch_key="Sample")此处我们预先对转录组数据进行了单独的批次校正、降维及聚类注释。这一步旨在建立一个单模态基准 (Baseline),以便后续与 MOFA+ 多模态整合分析的结果进行对比,评估引入甲基化数据是否提升了细胞分群的清晰度或生物学解释性。
#sc.pp.scale(adata_rna, max_value=10)
sc.tl.pca(adata_rna)if len(samples) > 1:
ho = run_harmony(
adata_rna.obsm['X_pca'],
meta_data=adata_rna.obs,
vars_use='Sample',
random_state=0,
max_iter_harmony=20)
adata_rna.obsm['X_pca_harmony'] = ho.Z_corr
rna_reduc_use = 'X_pca_harmony'
else:
rna_reduc_use = 'X_pca'
sc.pp.neighbors(adata_rna,use_rep = rna_reduc_use, random_state = 20)
sc.tl.umap(adata_rna, random_state = 20)
sc.tl.tsne(adata_rna,use_rep=rna_reduc_use, random_state = 20)
sc.tl.leiden(adata_rna, resolution=resolution, random_state=20)图注:本图展示了基于转录组数据的细胞聚类 UMAP 图。
- 左图:每个点代表一个细胞,颜色代表不同的细胞簇 (Leiden)。
- 右图:每个点代表一个细胞,颜色代表不同的样本 (Sample)。
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi=80)
sc.pl.umap(adata_rna, color = ['leiden','Sample'], wspace=0.3)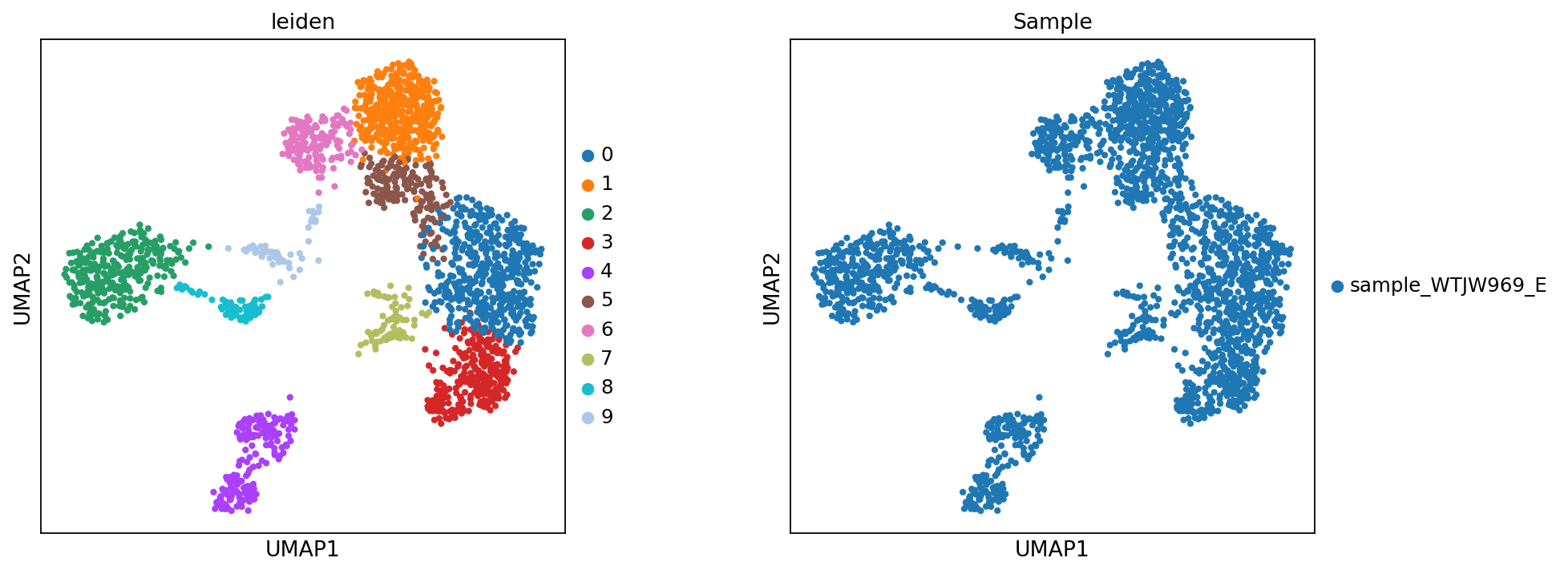
图注:本图展示了 RNA 数据在 UMAP 降维空间中特定标志基因的表达分布。
- 横坐标和纵坐标:UMAP 降维后的两个主维度(UMAP1 和 UMAP2)。
- 颜色:颜色深浅表示各标志基因的表达水平,颜色越深表示表达量越高。展示的标志基因包括:
CD3D、THEMIS(T 细胞)、NKG7(NK 细胞)、CD8A(CD8+ T 细胞)、CD4(CD4+ T 细胞)、CD79A和MS4A1(B 细胞)、JCHAIN(浆细胞)、LYZ(单核细胞)、LILRA4(pDC 细胞)。- 点:每个点代表一个单独的细胞。
- 用途:通过标志基因的表达模式,可以初步识别和验证不同细胞类型,辅助细胞类型注释。
colors = ["#F5E6CC", "#FDBB84", "#E34A33", "#7F0000"]
my_warm_cmap = mcolors.LinearSegmentedColormap.from_list("white_orange_red", colors, N=256)
marker_names = ["CD3D", "THEMIS", "NKG7", "CD8A", "CD4", "IL7R", "S100A4", "CCR7",
"CD79A","MS4A1","JCHAIN",
"LYZ","FCN1","FCGR3A","CST3","MKI67"]
sc.pl.umap(
adata_rna,
color=marker_names,
ncols=4,
color_map=my_warm_cmap,
vmax='p99',
s=10,
frameon=False,
vmin=0,
show=False
)
plt.gcf().set_size_inches(12, 8)
plt.show()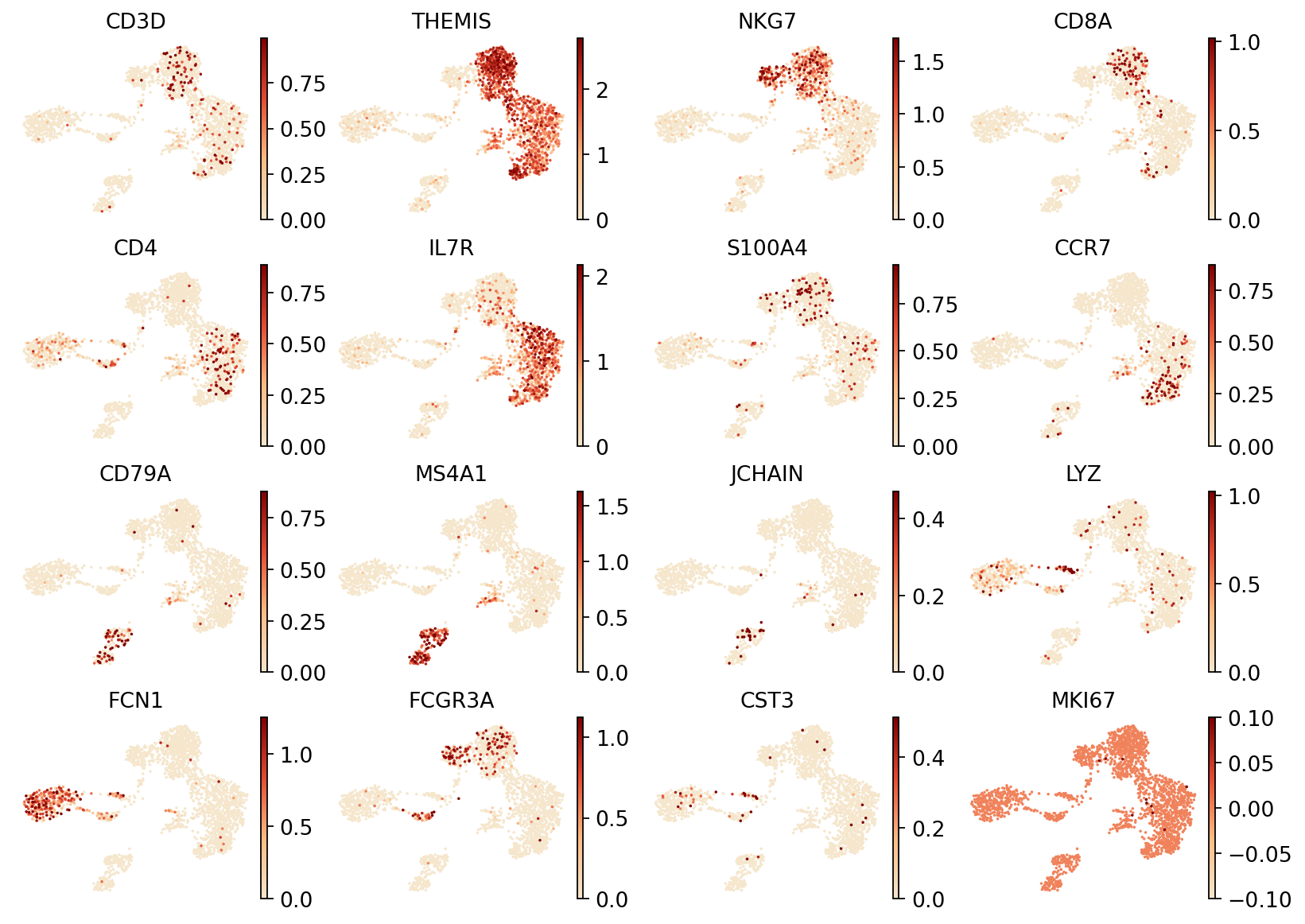
celltype = {
'0': 'Navie CD4+ T',
'1': 'CD8+ T',
'2': 'CD14+ Mono',
'3': 'Memory CD4+ T',
'4': 'B',
'5': 'T',
'6': 'NK',
'7': 'Multiplet',
'8': 'FCGR3A+ Mono',
'9': 'DC'
}
adata_rna.obs['celltype'] = adata_rna.obs['leiden'].map(celltype)if not 'celltype' in adata_rna.obs.columns:
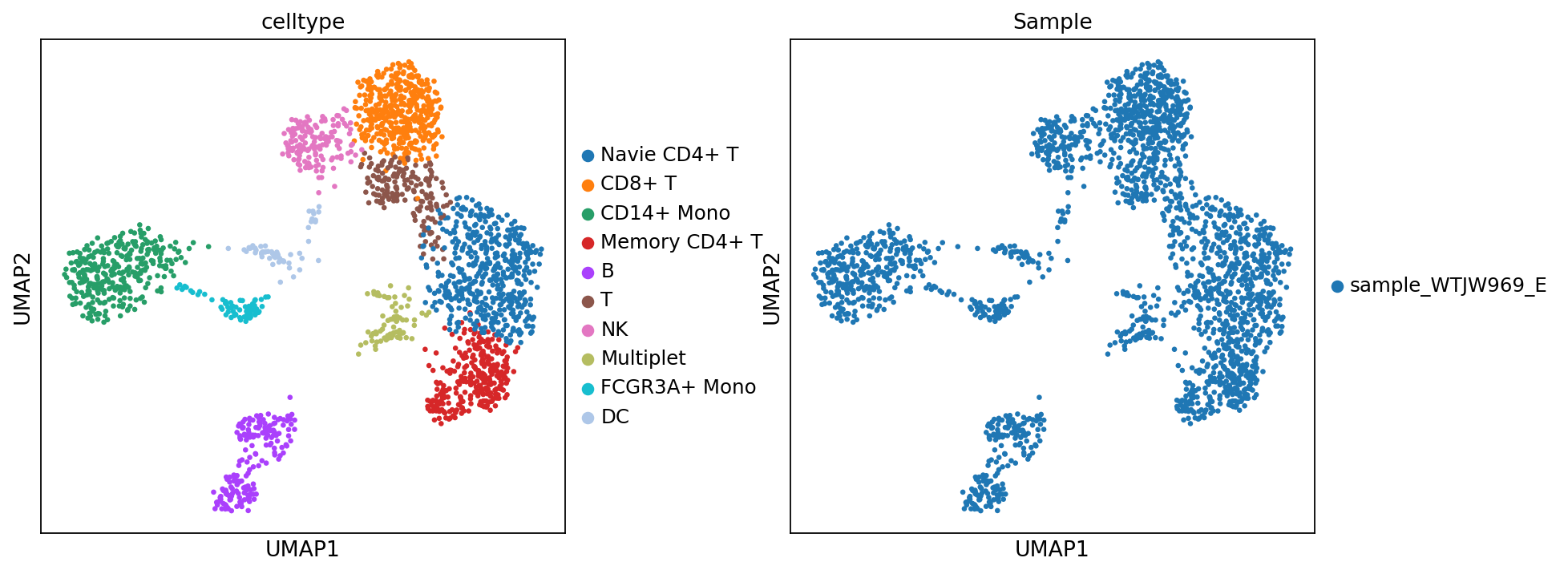
adata_rna.obs['celltype'] = adata_rna.obs['leiden']图注:本图展示了基于转录组数据注释后的细胞类型 UMAP 图。
- 左图:每个点代表一个细胞,颜色代表不同的细胞类型 (celltype)。
- 右图:每个点代表一个细胞,颜色代表不同的样本 (Sample)。
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi = 80)
sc.pl.umap(
adata_rna,
color=['celltype', 'Sample'],
ncols=2,
s=35,
wspace=0.3
)
构建 MuData 对象并训练 MOFA+ 模型
os.makedirs(f"{outdir}/models", exist_ok=True)mu_list = {}
mu_list['rna'] = adata_rna
for i in adata_met_list.keys():
mu_list[f'methy_{i}'] = adata_met_list[i]
mu_listobs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'mito', 'Sample', 'raw_Sample', 'resolution.0.5_d30', 'CellAnnotation', 'original_barcode', 'chemistry', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'leiden', 'celltype'
var: 'gene_ids', 'feature_types', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne', 'leiden', 'leiden_colors', 'Sample_colors', 'celltype_colors'
obsm: 'X_pca', 'X_umap', 'X_tsne'
varm: 'PCs'
layers: 'counts'
obsp: 'distances', 'connectivities',
'methy_geneslop2k': AnnData object with n_obs × n_vars = 2189 × 25405
obs: 'total_cpg_number', 'genome_cov', 'reads_counts', 'CpG%', 'CH%', 'Sample', 'original_barcode', 'chemistry'
var: 'chrom', 'end', 'start', 'cov_mean', 'mean', 'dispersion', 'cov', 'score', 'feature_select', 'highly_variable'}
构建多模态容器 (MuData) 与模型训练
MuData 是专为多组学数据设计的数据结构,可以同时存储和管理多个模态(如 RNA, Methylation)的数据及其关联。
MOFA+ 模型训练参数说明:
- groups_label:用于多样本/多批次分析,模型会尝试校正批次效应。
- n_factors:期望发现的潜在因子数量(默认 10)。因子数量越多,能捕捉的细节越多,但计算量增加且可能引入噪声。
- scale_views:是否对每个模态进行缩放。默认设为
False,以平衡不同模态的数据规模差异。 - convergence_mode:收敛模式,有
slow、medium、fast三种模式。slow模式训练更彻底,结果更稳定;fast模式速度更快。
# 构建 MuData 对象,整合 RNA 和 Methylation 数据
mdata = mu.MuData(mu_list)
mdata.obs['Sample'] = mdata['rna'].obs['Sample']
mdata.obs['celltype'] = mdata['rna'].obs['celltype']
mdata.obs['CpG%'] = mdata[mdata.mod_names[1]].obs['CpG%']
mdataMuData object with n_obs × n_vars = 2189 × 64011
obs: 'Sample', 'celltype', 'CpG%'
var: 'highly_variable'
2 modalities
rna: 2189 x 38606
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'mito', 'Sample', 'raw_Sample', 'resolution.0.5_d30', 'CellAnnotation', 'original_barcode', 'chemistry', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'leiden', 'celltype'
var: 'gene_ids', 'feature_types', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne', 'leiden', 'leiden_colors', 'Sample_colors', 'celltype_colors'
obsm: 'X_pca', 'X_umap', 'X_tsne'
varm: 'PCs'
layers: 'counts'
obsp: 'distances', 'connectivities'
methy_geneslop2k: 2189 x 25405
obs: 'total_cpg_number', 'genome_cov', 'reads_counts', 'CpG%', 'CH%', 'Sample', 'original_barcode', 'chemistry'
var: 'chrom', 'end', 'start', 'cov_mean', 'mean', 'dispersion', 'cov', 'score', 'feature_select', 'highly_variable'# 取多个模态细胞的交集,确保细胞一一对应
mu.pp.intersect_obs(mdata)
# 运行 MOFA+ 模型
mu.tl.mofa(mdata,
use_raw = False,
groups_label = 'Sample', # 指定样本分组列,用于处理批次效应
scale_views=True, # 平衡不同模态的权重
n_factors=10, # 设定潜在因子的数量
convergence_mode='slow', # 使用慢速模式以获得更稳健的结果
use_var = 'highly_variable', # 仅使用高变特征进行训练
outfile=f"{outdir}/models/pbmc_969_multi_methy.hdf5") # 模型保存路径 #########################################################
### __ __ ____ ______ ###
### | \/ |/ __ \| ____/\ _ ###
### | \ / | | | | |__ / \ _| |_ ###
### | |\/| | | | | __/ /\ \_ _| ###
### | | | | |__| | | / ____ \|_| ###
### |_| |_|\____/|_|/_/ \_\ ###
### ###
#########################################################
Scaling views to unit variance...
Loaded view='rna' group='sample_WTJW969_E' with N=2189 samples and D=2000 features...
Loaded view='methy_geneslop2k' group='sample_WTJW969_E' with N=2189 samples and D=2500 features...
Model options:
- Automatic Relevance Determination prior on the factors: True
- Automatic Relevance Determination prior on the weights: True
- Spike-and-slab prior on the factors: False
- Spike-and-slab prior on the weights: True
Likelihoods:
- View 0 (rna): gaussian
- View 1 (methy_geneslop2k): gaussian
######################################
## Training the model with seed 1 ##
######################################
Converged!
#######################
## Training finished ##
#######################
Warning: Output file /home/weiqiuxia/workspace/project/weiqiuxia/MOFA_results/models/pbmc_969_multi_methy.hdf5 already exists, it will be replaced
Saving model in /home/weiqiuxia/workspace/project/weiqiuxia/MOFA_results/models/pbmc_969_multi_methy.hdf5...
Saved MOFA embeddings in .obsm['X_mofa'] slot and their loadings in .varm['LFs'].
mdata.uns = mdata.uns or dict()MOFA 因子可视化
MOFA+ 因子分析的核心结果之一是推断出的潜在因子。我们可以通过可视化这些因子在细胞中的分布,来理解不同细胞类型的生物学差异。
图注:本图(散点图矩阵)展示了前 10 个 MOFA 因子(两两配对)在细胞中的分布。
- 颜色:代表细胞类型 (
rna:celltype) 或甲基化水平 (methy_geneslop2k:CpG%)。- 观察点:如果某个因子(如 Factor 1)能清晰地将某种细胞类型(如 B cells)与其他细胞分开,说明该因子捕捉到了该细胞类型的特异性生物学信号。
mu.pl.mofa(mdata, color="celltype", components=["1,2", "3,4", "5,6", "7,8", "9,10"])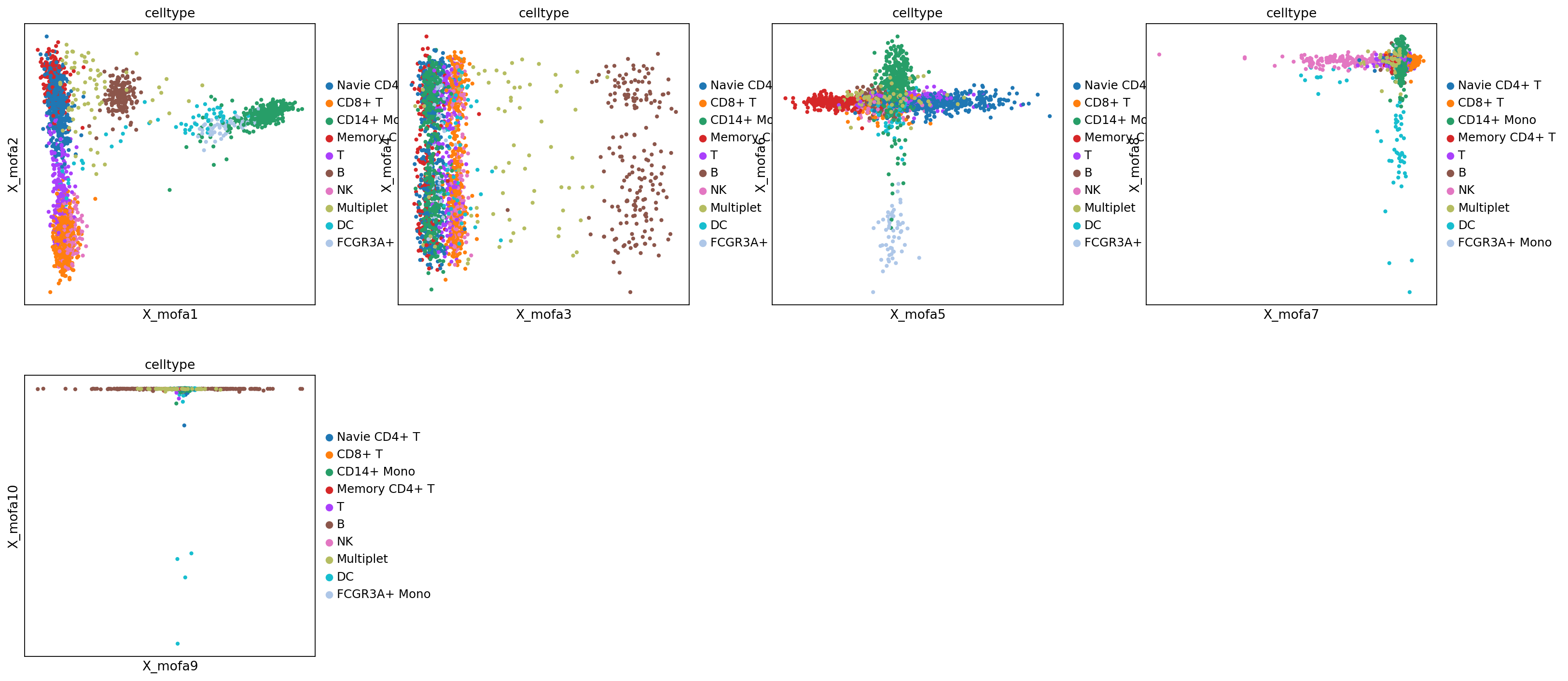
mu.pl.mofa(mdata, color="CpG%", components=["1,2", "3,4", "5,6", "7,8","9,10"])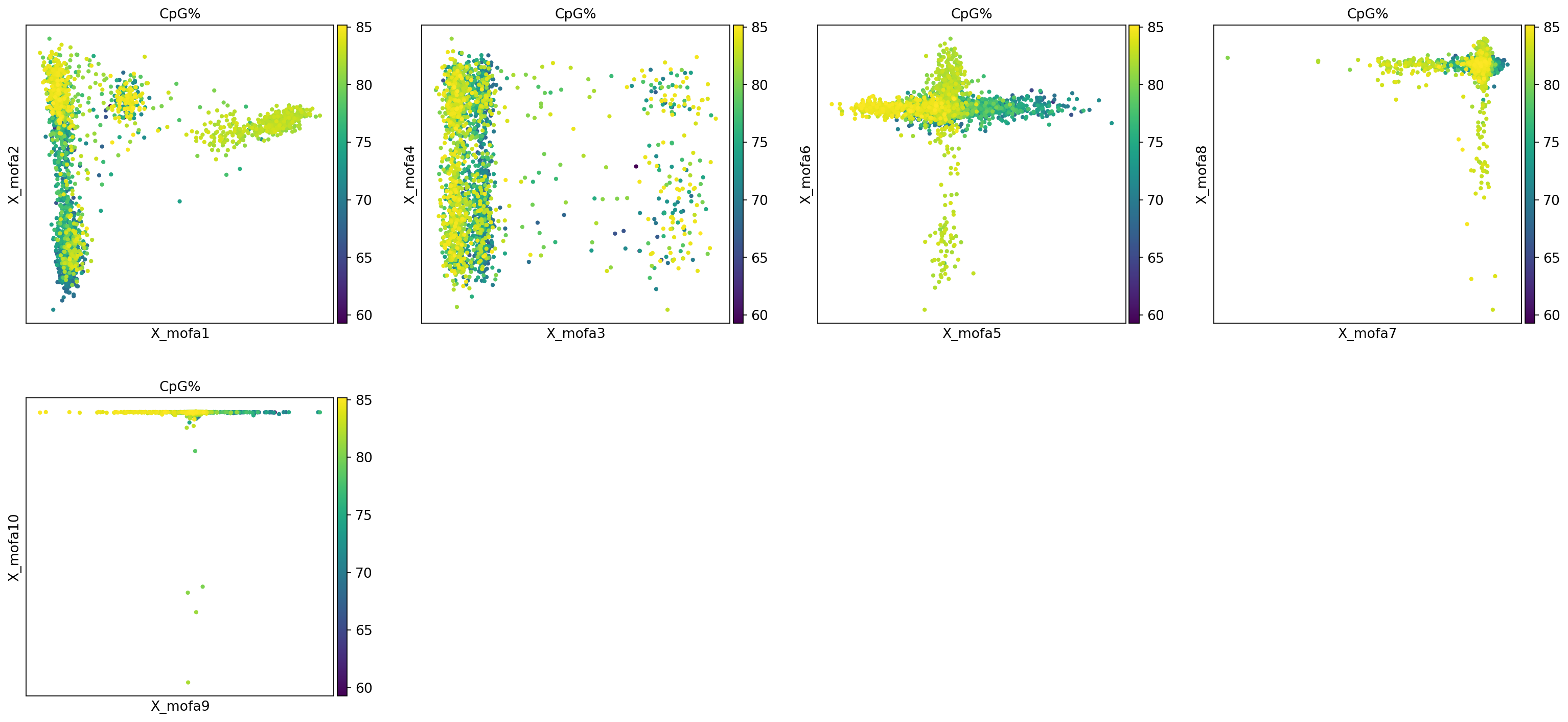
模型评估与方差解释 (R2)
评估每个因子对不同组学模态变异的解释度 (Variance Explained, R2)。
model = mfx.mofa_model(f"{outdir}/models/pbmc_969_multi_methy.hdf5")
modelSamples (cells): 2189
Features: 4500
Groups: sample_WTJW969_E (2189)
Views: methy_geneslop2k (2500), rna (2000)
Factors: 10
Expectations: W, Z
model.get_r2()| Factor | View | Group | R2 | |
|---|---|---|---|---|
| 0 | Factor1 | rna | sample_WTJW969_E | 1.755070e+01 |
| 1 | Factor1 | methy_geneslop2k | sample_WTJW969_E | 1.526547e-06 |
| 2 | Factor2 | rna | sample_WTJW969_E | 6.230776e+00 |
| 3 | Factor2 | methy_geneslop2k | sample_WTJW969_E | 1.247076e-05 |
| 4 | Factor3 | rna | sample_WTJW969_E | 5.786080e+00 |
| 5 | Factor3 | methy_geneslop2k | sample_WTJW969_E | 6.985795e-07 |
| 6 | Factor4 | rna | sample_WTJW969_E | 5.757473e-06 |
| 7 | Factor4 | methy_geneslop2k | sample_WTJW969_E | 2.565718e+00 |
| 8 | Factor5 | rna | sample_WTJW969_E | 1.429741e+00 |
| 9 | Factor5 | methy_geneslop2k | sample_WTJW969_E | 1.103752e-04 |
| 10 | Factor6 | rna | sample_WTJW969_E | 9.622432e-01 |
| 11 | Factor6 | methy_geneslop2k | sample_WTJW969_E | 7.399176e-06 |
| 12 | Factor7 | rna | sample_WTJW969_E | 8.270760e-01 |
| 13 | Factor7 | methy_geneslop2k | sample_WTJW969_E | 2.246223e-06 |
| 14 | Factor8 | rna | sample_WTJW969_E | 5.685163e-01 |
| 15 | Factor8 | methy_geneslop2k | sample_WTJW969_E | 3.142382e-06 |
| 16 | Factor9 | rna | sample_WTJW969_E | 4.485491e-01 |
| 17 | Factor9 | methy_geneslop2k | sample_WTJW969_E | 2.246235e-05 |
| 18 | Factor10 | rna | sample_WTJW969_E | 8.303199e-02 |
| 19 | Factor10 | methy_geneslop2k | sample_WTJW969_E | -2.510329e-07 |
r2_df = model.get_r2()
r2_df[r2_df['R2'] >= 1]| Factor | View | Group | R2 | |
|---|---|---|---|---|
| 0 | Factor1 | rna | sample_WTJW969_E | 17.550695 |
| 2 | Factor2 | rna | sample_WTJW969_E | 6.230776 |
| 4 | Factor3 | rna | sample_WTJW969_E | 5.786080 |
| 7 | Factor4 | methy_geneslop2k | sample_WTJW969_E | 2.565718 |
| 8 | Factor5 | rna | sample_WTJW969_E | 1.429741 |
图注:本热图展示了每个 MOFA 因子 (Factor) 对不同组学模态 (View) 变异的解释能力。
- 横轴:MOFA 因子 (Factor 1, 2, ...)。
- 纵轴:组学模态 (RNA, Methylation)。
- 颜色深浅:R2 值(方差解释度),颜色越深代表该因子对该模态的贡献越大。
- 意义:若某因子在两个模态均深色,通常为共享因子;若仅在一个模态深色,通常为私有因子。
mfx.plot_r2(model, x='View', vmax=10)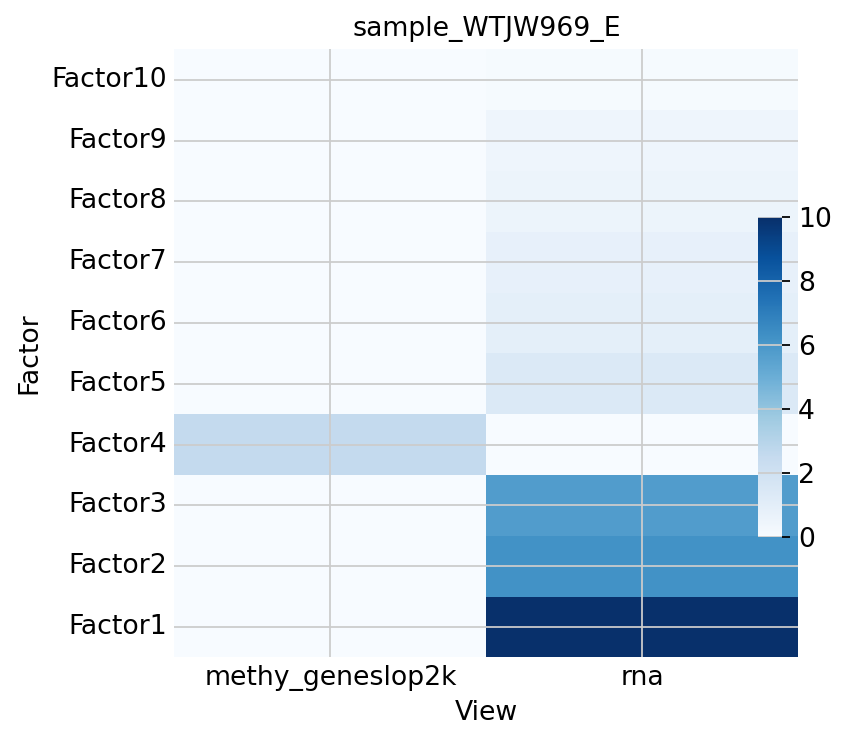
model.close()基于 MOFA 因子的下游分析
利用 MOFA 提取的潜在因子 (X_mofa) 替代原始特征,构建邻居图并进行 UMAP 降维和 Leiden 聚类。结合转录组的注释结果,评估是否能获得更清晰的细胞图谱。
在此步骤中,我们通常选择方差解释度较高的因子用于后续分析。
sc.pp.neighbors(mdata, use_rep="X_mofa", n_pcs=5, random_state=20)
sc.tl.umap(mdata, min_dist=.2, spread=1., random_state=20)
sc.tl.leiden(mdata, resolution=.5, random_state=20)图注:本图展示了基于 MOFA 因子构建的 UMAP 降维图。
- 此图综合了 RNA 和 Methylation 的信息,通常比单模态聚类更能区分细胞亚群。
- 颜色标识:
celltype:基于转录组数据的细胞类型注释。leiden:基于 MOFA 因子的聚类结果。CpG%:全基因组 CpG 甲基化水平。Sample:样本来源。
sc.pl.umap(mdata, color=["celltype","leiden",'CpG%', 'Sample'],legend_loc="on data", legend_fontoutline=1)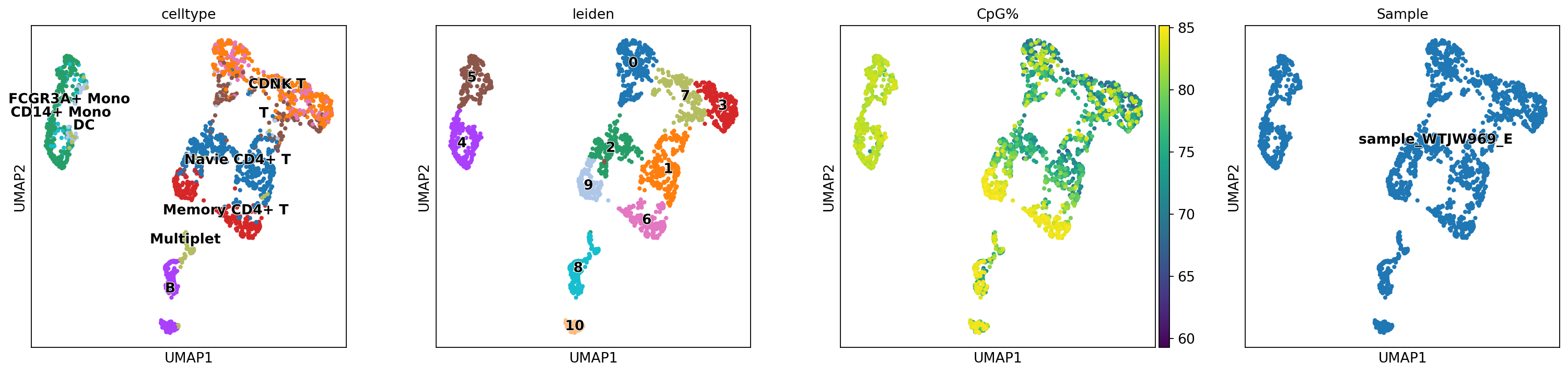
结果讨论与优化建议
MOFA+ 是基于高变特征进行后续的多模态整合分析的,因此它受筛选出的高变特征集影响较大。如果筛选出的高变特征集本身无法正确区分不同的生物学细胞群,那么 MOFA+ 的整合结果也会受到限制。
如上图所示,如果发现 MOFA+ 的多模态整合分群效果不如单组学(如仅 RNA)的分群结果,一个可能的原因是我们使用的甲基化区域粒度“太大”(例如 geneslop2k 指的是基因区域加上它上下游 2kb)。已有文献表明,基因不同区域(如启动子、增强子、基因体)的甲基化水平具有不同的调控意义。粗粒度的区域可能掩盖了精细的生物学信号,导致筛选出的高变特征集包含较多噪声,从而影响 MOFA+ 的整合效果。
优化策略: 已有利用 MOFA+ 整合甲基化和转录组数据的成功案例表明,精细筛选甲基化特征集至关重要。例如,在文献 Multi-omics profiling of mouse gastrulation at single-cell resolution 中,研究者采取了以下策略:
- 特征预筛选:利用公共数据或 ChIP-seq 数据,先定位与样本类型相关的关键调控区域,如
H3K27ac(Enhancers)、H3K4me3(TSS) 以及蛋白编码基因转录起始位点上下游 2kb 区域 (Promoters)。 - 高变筛选:基于这些精选的区域再进行高变特征筛选。
- 因子发现:在甲基化模态上找出与生物学过程强相关的因子。
通过这种方式,可以显著提高输入特征的信噪比,从而提升 MOFA+ 整合分析的效果。
输出文件与后续复用
分析完成后,我们将三个对象写入 outdir:
adata_rna.h5ad:RNA 的AnnData对象。adata_met_**.h5ad:甲基化的AnnData对象。mdata.h5mu:多组学MuData对象。
注意:
.h5mu文件通常较大,建议放在稳定的路径并做好备份。- 如果您改变了过滤阈值或
var_dim参数,建议重新运行完整流程并覆盖输出文件。
adata_rna.write_h5ad(f'{outdir}/adata_rna.h5ad')
for i in adata_met_list.keys():
adata_met_list[i].write_h5ad(f'{outdir}/adata_met_{i}.h5ad')
mdata.write(f'{outdir}/mdata.h5mu')更多资源