scATAC-seq Multi-Sample Integration Tutorial (Signac / LSI)
Environment Preparation
Loading R Packages
Please select the common_r environment for this integration tutorial
# Load necessary R packages
suppressPackageStartupMessages({
library(Seurat)
library(Signac)
library(EnsDb.Hsapiens.v86, lib.loc = "/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/python3.10/lib/R/library")
library(BSgenome.Hsapiens.UCSC.hg38,lib = "/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/python3.10/lib/R/library")
library(biovizBase, lib = "/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/python3.10/lib/R/library")
#library(BSgenome.Mmusculus.UCSC.mm10)
#library(EnsDb.Mmusculus.v79)
library(dplyr)
library(ggplot2)
library(patchwork)
library(harmony)
})
# Set random seed
set.seed(1234)
# Note, if data is large, you can customize resource settings
options(future.globals.maxSize = 15 * 1024^3) # 15GBmy36colors <-c( '#E5D2DD', '#53A85F', '#F1BB72', '#F3B1A0', '#D6E7A3', '#57C3F3', '#476D87',
'#E95C59', '#E59CC4', '#AB3282', '#23452F', '#BD956A', '#8C549C', '#585658',
'#9FA3A8', '#E0D4CA', '#5F3D69', '#C5DEBA', '#58A4C3', '#E4C755', '#F7F398',
'#AA9A59', '#E63863', '#E39A35', '#C1E6F3', '#6778AE', '#91D0BE', '#B53E2B',
'#712820', '#DCC1DD', '#CCE0F5', '#CCC9E6', '#625D9E', '#68A180', '#3A6963',
'#968175', "#6495ED", "#FFC1C1",'#f1ac9d','#f06966','#dee2d1','#6abe83','#39BAE8','#B9EDF8','#221a12',
'#b8d00a','#74828F','#96C0CE','#E95D22','#017890')Get Gene Annotation Information
We will obtain genomic annotation information (gene locations, transcripts, exons, TSS, etc.) for the corresponding species from the EnsDb database. The specific species needs to be changed according to the data. This information is used for:
- Calculate TSS Enrichment for ATAC (Judge if open chromatin is more concentrated near Transcription Start Sites)
- Building Gene Activity Matrix (Mapping peaks signals to genes)
- Peak Annotation and Functional Analysis
Notes:
- Species matches reference genome version (e.g.,
EnsDb.Hsapiens.v86for hg38,EnsDb.Mmusculus.v75for mm10) - Consistent chromosome naming style (e.g., using
chr1,chr2prefixes)
# This tutorial uses human data, so we get gene annotation information
suppressWarnings({
suppressMessages({
annotation <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
seqlevels(annotation) <- paste0('chr', seqlevels(annotation))
genome(annotation) <- 'hg38'
})
})
# Set parallel computing (silent processing, optional)
suppressPackageStartupMessages({
library(future)
})
plan("multicore", workers = 4)Data Reading
This tutorial provides two different forms of data input to meet the data acquisition needs of different users. Just choose the one that suits you:
Cloud Platform RDS File Reading
Data Characteristics:
- RDS file is a standard Seurat object file
- Data has been preprocessed and multiple samples merged
- Can be directly used for subsequent downstream analysis, or extraction of expression matrices for reintegration
Applicable Scenarios:
- When you cannot obtain the standard
filtered_peaks_bc_matrixexpression matrix and fragment matrix - When you want to integrate existing cloud platform data for learning
- When you need to quickly re-perform scRNA-seq data integration analysis
Notes:
- For specific data mounting and RDS file reading, please refer to the Jupyter usage tutorial
For example, the following project data /home/demo-SeekGene-com/workspace/data/AY1752565399550/
library(Seurat)
library(Signac)
# 1. Read Data
input <- readRDS("/home/demo-seekgene-com/workspace/data/AY1752565399550/input.rds")
meta <- read.table("/home/demo-seekgene-com/workspace/data/AY1752565399550/meta.tsv",
header = TRUE,
sep = "\t",
row.names = 1)
# 2. Extract ATAC data and genome annotation
counts <- input@assays$ATAC@counts # ATAC count matrix
fragments <- input@assays$ATAC@fragments # Fragment file (if available)
annotations <- input@assays$ATAC@annotation # Genome annotation
# 3. Create ChromatinAssay (Signac format ATAC data)
atac_assay <- CreateChromatinAssay(
counts = counts,
fragments = fragments,
annotation = annotations
)
# 4. Create Seurat object (containing only ATAC data)
atac_seurat <- CreateSeuratObject(
counts = atac_assay, # Use ChromatinAssay as input
assay = "ATAC", # Specify assay name
meta.data = meta # Add sample metadata
)
seurat_list <- SplitObject(atac_seurat, split.by = "Sample")
# 6. Clean up memory
rm(input, counts, fragments, annotations, atac_assay)
gc()Standard filtered_peaks_bc_matrix File Reading
Applicable Scenarios:
- When you have standard gene expression matrices and peaks open matrix files
- When you want to independently complete multi-sample integration and batch correction of single-cell multi-omics (SeekArc) data
- When a complete workflow from raw data to integration analysis is needed
Note:
- Please ensure the file structure between samples is as follows:
The data directory structure must meet the following requirements:
- The folder name for each sample is the Sample ID, such as S127, S44R, etc.
- Each sample folder contains the following files:
filtered_peaks_bc_matrix: ATAC peak open matrix folder, containingbarcodes.tsv.gz,features.tsv.gz, andmatrix.mtx.gzfiles.{Sample ID}_A_fragments.tsv.gz: ATAC fragments file, such as S127_A_fragments.tsv.gz.{Sample ID}_A_fragments.tsv.gz.tbi: ATAC fragments index file, such as S127_A_fragments.tsv.gz.tbi.
The specific folder structure is as follows:
scATAC-seq Data (Peaks Matrix + Fragments)
├── S127/
│ ├── filtered_peaks_bc_matrix/
│ │ ├── barcodes.tsv.gz (Cell barcodes)
│ │ ├── features.tsv.gz (Peaks list)
│ │ └── matrix.mtx.gz (Sparse count matrix)
│ ├── S127_A_fragments.tsv.gz (Genomic coordinates of each sequencing read)
│ ├── S127_A_fragments.tsv.gz.tbi (Index file for fragments)
│ └── per_barcode_metrics.csv (Optional, per-cell QC metrics)
├── S44R/
│ ├── filtered_peaks_bc_matrix/
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── S44R_A_fragments.tsv.gz
│ ├── S44R_A_fragments.tsv.gz.tbi
│ └── per_barcode_metrics.csv (optional)
# Define sample names
sample_names <- c('S127', 'S44R')
# Create an empty list to store the Seurat object for each sample
seurat_list <- list()
for (sample in sample_names) {
cat('Processing sample:', sample, '\n')
# Construct file paths
counts_path <- file.path(sample, 'filtered_peaks_bc_matrix')
fragments_path <- file.path(sample, paste0(sample, '_A_fragments.tsv.gz'))
#metadata_path <- file.path(sample, 'per_barcode_metrics.csv')
# Read peaks matrix data
counts <- Read10X(counts_path)
# Read QC metrics metadata
#metadata <- read.csv(
# file = metadata_path,
# header = TRUE,
# row.names = 1
#)
# Creating ChromatinAssay object
chrom_assay <- CreateChromatinAssay(
counts = counts,
sep = c(":", "-"),
fragments = fragments_path,
min.cells = 3,
min.features = 200
)
# Create Seurat object
obj <- CreateSeuratObject(
counts = chrom_assay,
assay = "ATAC"#,meta.data = metadata
)
# Add gene annotation information
Annotation(obj) <- annotation
# Add sample ID
obj$Sample <- sample
obj$orig.ident <- sample
# Adding Seurat object to list
seurat_list[[sample]] <- obj
cat('Sample', sample, 'processed, cell count:', ncol(obj), '\n')
}Quality Control
QC Metrics Calculation
Common scATAC-seq QC metrics:
TSS.enrichment(TSS Enrichment Score, usually > 2): Higher is better, indicating signals are more concentrated near transcription start sites; too low may indicate high background or poor cell viability.nucleosome_signal(Nucleosome Signal, lower is better): Reflects nucleosome periodicity signal; usually lower is better.blacklist_ratio(Blacklist Ratio): Proportion of reads falling into genomic blacklist regions; too high indicates data quality issues.nCount_ATAC: Total ATAC counts; extremely low or high values require caution.
Actual thresholds should be set based on data distribution and judged in combination with fragments quality, doublet ratios, etc.
# Quality control for each sample
suppressWarnings({
suppressMessages({
for (sample_name in names(seurat_list)) {
seurat_obj <- seurat_list[[sample_name]]
# Set default assay to ATAC
DefaultAssay(seurat_obj) <- 'ATAC'
# Calculate TSS enrichment score
seurat_obj <- TSSEnrichment(seurat_obj)
# Calculate nucleosome signal
seurat_obj <- NucleosomeSignal(seurat_obj)
# Calculate blacklist region read ratio
seurat_obj$blacklist_ratio <- FractionCountsInRegion(
object = seurat_obj,
assay = 'ATAC',
regions = blacklist_hg38_unified)
# Update objects in seurat_list
seurat_list[[sample_name]] <- seurat_obj
}
})
})Extracting fragments at TSSs
Computing TSS enrichment score
Extracting TSS positions
Extracting fragments at TSSs
Computing TSS enrichment score
QC Metrics Visualization
Use violin plots to view the distribution/outliers of each QC metric to determine appropriate thresholds:
Suggestion:
- Observe whether there is an obvious long tail or bimodal distribution
- Try multiple thresholds and compare if downstream clustering/UMAP is clearer
# Visualize QC metrics (optional execution)
options(repr.plot.width = 17, repr.plot.height = 10)
suppressWarnings({
for (sample_name in names(seurat_list)) {
seurat_obj <- seurat_list[[sample_name]]
p1=DensityScatter(seurat_obj, x = 'nCount_ATAC', y = 'TSS.enrichment', log_x = TRUE, quantiles = TRUE)
p2=VlnPlot(
object = seurat_obj,
features = c('nCount_ATAC', 'TSS.enrichment', 'blacklist_ratio', 'nucleosome_signal'),
pt.size = 0.1,
ncol = 5
)
print(p1 / p2 + plot_annotation(title = sample_name))
}
})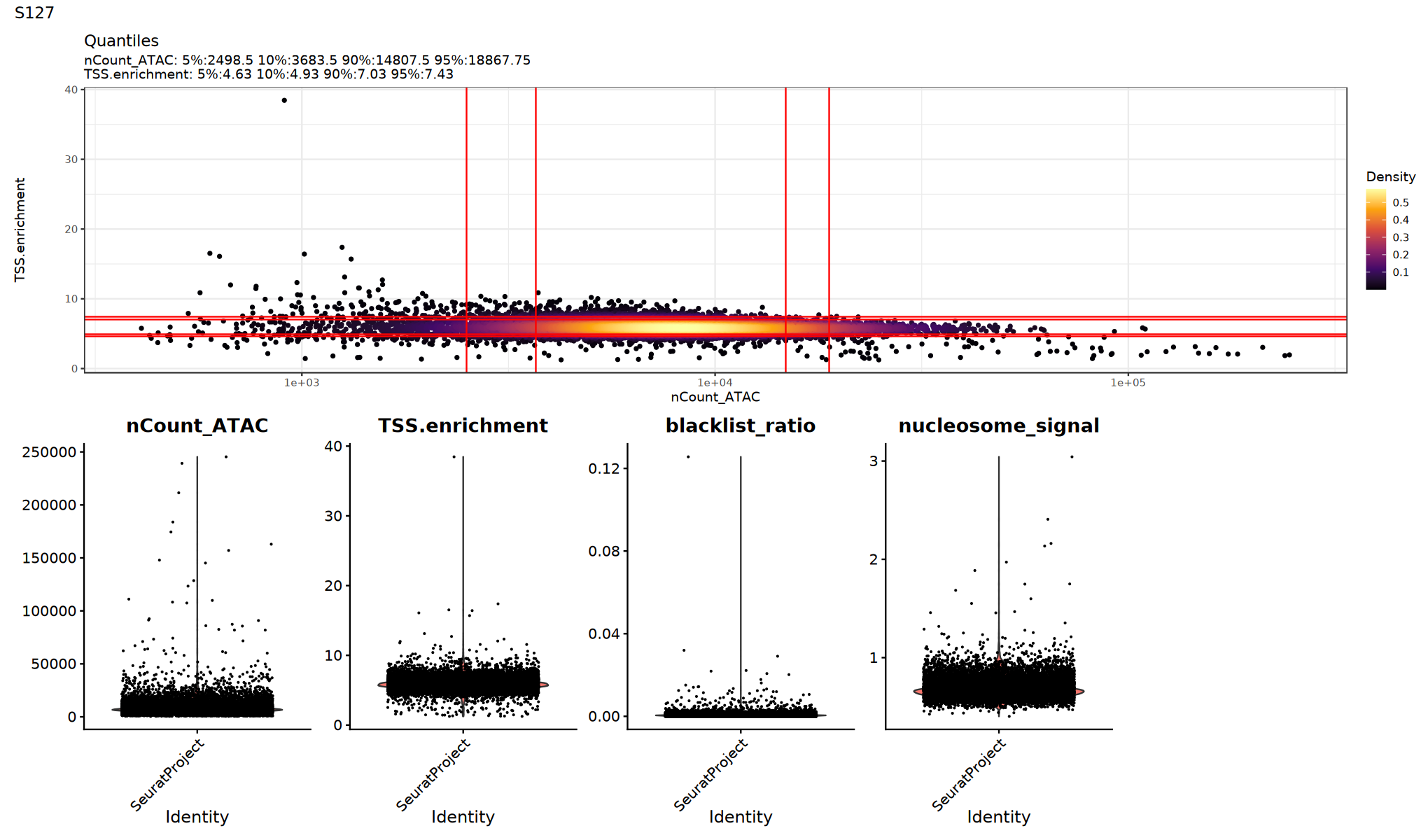
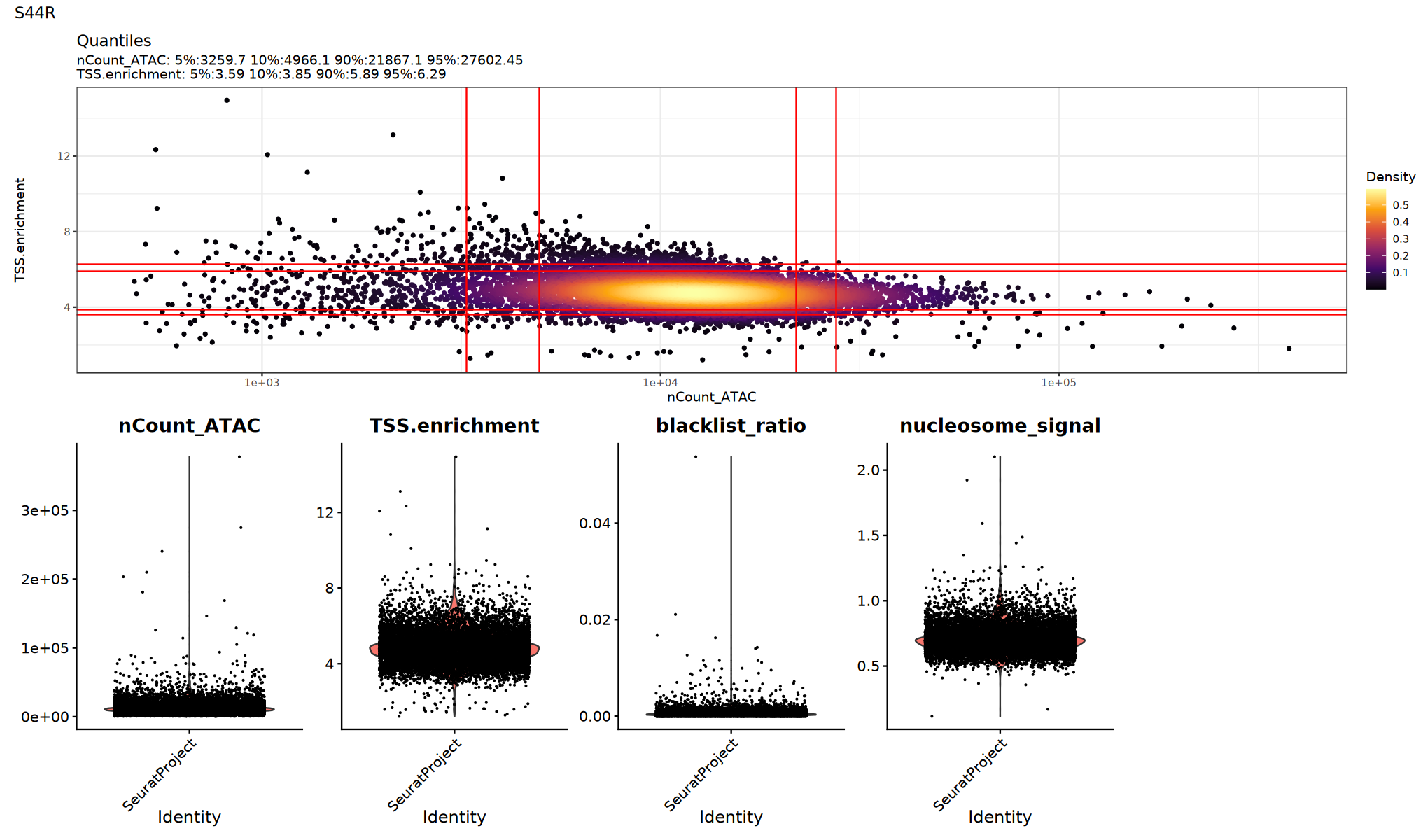
Low Quality Cell Filtering
Filter low-quality cells based on QC metrics. Specific thresholds should be adjusted according to data characteristics. Refer to the violin plot distribution above for specific filtering thresholds.
# Set QC filtering thresholds (adjust based on data characteristics)
for (sample_name in names(seurat_list)) {
cat('Filtering sample', sample_name, '...\n')
seurat_obj <- seurat_list[[sample_name]]
# Record cell count before filtering
cells_before <- ncol(seurat_obj)
# Filter based on QC metrics
seurat_obj <- subset(
x = seurat_obj,
subset = nCount_ATAC > 500 &
nCount_ATAC < 100000 &
blacklist_ratio < 0.05 &
nucleosome_signal < 2 &
TSS.enrichment > 1
)
# Record cell count after filtering
cells_after <- ncol(seurat_obj)
# Update seurat_list
seurat_list[[sample_name]] <- seurat_obj
cat('Sample', sample_name, 'filtered: ', cells_before, ' cells before, ', cells_after, ' cells after\n')
}正在对样本 S44R 进行质控过滤...n 样本 S44R 过滤完成: 过滤前 12068 个细胞,过滤后 12050 个细胞
Calculate Common Peaks Between Samples
Since peak calling is performed independently for each sample, the peaks in different samples are different. To integrate multiple samples, we need to unify peak information and create a common peak set for all samples.
Processing Workflow:
- Merge peaks from all samples
- Filter peak length (20bp-10kb)
- Re-quantify common peaks for each sample
Notes: If the first method (reading RDS) was used to read data earlier, this part does not need to be analyzed.
Notes: If the first method (reading RDS) was used to read data earlier, this part does not need to be analyzed.
Notes: If the first method (reading RDS) was used to read data earlier, this part does not need to be analyzed.
# Extract peaks information for each sample
all_peaks <- lapply(seurat_list, function(x) {
DefaultAssay(x) <- "ATAC"
granges(x@assays$ATAC)
})
# Merge peaks from all samples
gr_list <- GRangesList(all_peaks)
all_granges <- unlist(gr_list, use.names = FALSE)
combined.peaks <- reduce(x = all_granges)
# Filter peak length (keep 20bp-10kb peaks)
peakwidths <- width(combined.peaks)
common_peaks <- combined.peaks[peakwidths < 10000 & peakwidths > 20]
cat('Total peaks count:', length(common_peaks), '\n')
# Quantify common peaks for each sample
seurat_list <- lapply(seurat_list, function(x) {
cat('Quantifying common peaks for sample', x$Sample[1], '...\n')
# Quantify counts matrix for common peaks
combined_counts <- FeatureMatrix(
fragments = Fragments(x@assays$ATAC),
features = common_peaks,
cells = colnames(x)
)
# Create new ChromatinAssay
combined_peaks_assay <- CreateChromatinAssay(
counts = combined_counts,
fragments = Fragments(x@assays$ATAC),
annotation = Annotation(x@assays$ATAC)
)
# Add to Seurat object
x[["combinedpeaks"]] <- combined_peaks_assay
DefaultAssay(x) <- "combinedpeaks"
x[["ATAC"]] <- NULL
return(x)
})正在为样本 S127 量化共有peaks...n
Extracting reads overlapping genomic regions
正在为样本 S44R 量化共有peaks...n
Extracting reads overlapping genomic regions
Multi-Sample Merging
In the multi-omics data integration analysis workflow, multi-sample merging is an important prerequisite step for integration analysis.
Purpose of Merging:
- Integrate data from multiple samples into a single Seurat object
- Prepare for subsequent batch correction and integration analysis
- Facilitate comparative analysis with batch-corrected results
Important Note:
- This step only performs simple data merging, batch effect correction has not been performed yet
- Merged object contains raw data from all samples
- Perform normalization, feature selection, PCA dimensionality reduction, and UMAP visualization on merged data
suppressWarnings({
suppressMessages({
# Merge scATAC-seq datasets
obj.combined <- merge(seurat_list[[1]], seurat_list[-1], merge.data = FALSE)
# Set default assay and normalize
DefaultAssay(obj.combined) <- "combinedpeaks"
obj.combined <- FindTopFeatures(obj.combined, min.cutoff = 10)
obj.combined <- RunTFIDF(obj.combined)
obj.combined <- RunSVD(obj.combined)
obj.combined <- RunUMAP(obj.combined, reduction = "lsi", dims = 2:30,reduction.name="atacumap")
})
})Data Integration
Now we will integrate scATAC-seq data from multiple samples. There are two common methods for scATAC-seq data integration:
Integration Method Selection
CCA (Canonical Correlation Analysis) Integration:
- Integration method based on Canonical Correlation Analysis (CCA)
- Integrate by finding common variation patterns between samples
- Suitable for cases with large differences between samples
- Classic integration method in Seurat package
Harmony Integration:
- Fast integration method based on iterative clustering
- Correct batch effects directly in dimensionality reduction space
- High computational efficiency, suitable for large-scale data
- Retain more biological variation information
Method Selection Suggestions
- Harmony: scATAC-seq data from the same platform but different samples; Harmony is faster for large numbers of samples or large datasets.
- CCA: Runs longer, usually recommended when batch effects are severe, such as cross-platform scATAC-seq data.
Integration using Harmony
Note: Choose either Harmony or CCA for batch correction
atac_integrate_harmony <- function(obj) {
library(harmony)
DefaultAssay(obj) <- "combinedpeaks"
obj<- RunHarmony(
object = obj,
group.by.vars = 'Sample',
reduction = 'lsi',
assay.use = 'combinepeaks',
reduction.save = "harmonylsi",
project.dim = FALSE
)
obj <- RunUMAP(obj,reduction = 'harmonylsi',reduction.name="atacharmonyumap", dims = 2:30)
#obj <- RunTSNE(obj, reduction = "harmonylsi",reduction.name="atacharmonytsne",dims = 2:50,check_duplicates = FALSE)
return(obj)
}
suppressWarnings({
suppressMessages({
integrated=atac_integrate_harmony(obj.combined)
integrated <- FindNeighbors(object = integrated, reduction = 'harmonylsi', graph.name = "atacneighbor", dims = 2:30)
integrated <- FindClusters(object = integrated, verbose = FALSE, graph.name = "atacneighbor",resolution = 0.5, algorithm = 3)
})
})
# Visualize ATAC batch correction effect
options(repr.plot.width=10, repr.plot.height=8)
DimPlot(integrated, reduction = "atacharmonyumap", group.by = "Sample")Integration using CCA
Note: Choose either Harmony or CCA for batch correction
# Define ATAC integration function
options(future.globals.maxSize = 20 * 1024^3)
integrate_atac_cca <- function(object.list){
object.list <- lapply(object.list, function(x) {
DefaultAssay(x)="combinedpeaks"
x=RunTFIDF(x)
x=FindTopFeatures(x, min.cutoff = 5)
x=RunSVD(x)
return(x)
})
anchors <- FindIntegrationAnchors(
object.list = object.list,
anchor.features = rownames(obj.combined),
reduction = "rlsi",
dims = 2:30
)
integrated <- IntegrateEmbeddings(
anchorset = anchors,
reductions = obj.combined[["lsi"]],
new.reduction.name = "integrated_lsi",
dims.to.integrate = 1:30)
integrated<- RunUMAP(integrated, reduction = "integrated_lsi", dims = 2:30,reduction.name="atacintegratedumap")
#integrated<- RunTSNE(integrated, reduction = "integrated_lsi", dims = 2:30,reduction.name="atacintegratedtsne")
return(integrated)
}
# Execute ATAC integration
suppressWarnings({
suppressMessages({
integrated <- integrate_atac_cca(seurat_list)
integrated <- FindNeighbors(object = integrated, reduction = 'integrated_lsi', graph.name = "atacneighbor", dims = 2:30)
integrated <- FindClusters(object = integrated, verbose = FALSE, graph.name = "atacneighbor",resolution = 0.5, algorithm = 3)
})
})
# Visualize ATAC batch correction effect
options(repr.plot.width=10, repr.plot.height=8)
DimPlot(integrated, reduction = "atacintegratedumap", group.by = "Sample")开始处理各样本的LSI降维...n 寻找整合锚点...n 整合嵌入向量...
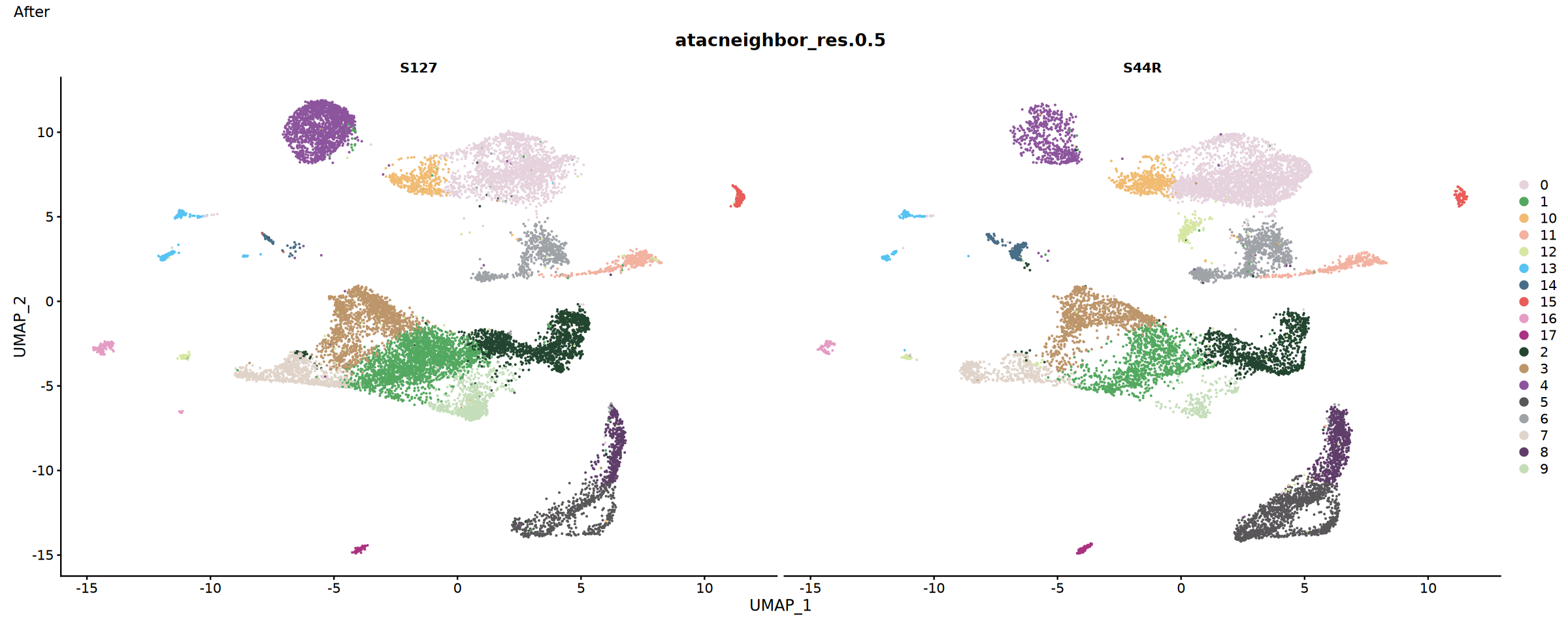
Integration Effect Assessment
Compare results before and after integration to assess integration effect.
Assessment Metrics:
- Sample mixing degree: Distribution of cells from different samples in UMAP plot
- Batch effect removal: Clustering of technical replicates
- Biological signal preservation: Separation of known cell types
suppressWarnings({
suppressMessages({
# Create control data before integration
obj.combined <- FindNeighbors(object = obj.combined,
reduction = 'lsi',
graph.name = "atacneighbor",
dims = 2:30)
# Cluster Analysis
obj.combined <- FindClusters(object = obj.combined,
verbose = FALSE,
graph.name = "atacneighbor",
cluster.name = "atac_clusters",
resolution = 0.5,
algorithm = 3)
})
})options(repr.plot.width=15, repr.plot.height=6)
# Sample distribution before integration
p1 <- DimPlot(obj.combined, split.by = "Sample" , cols = my36colors)+plot_annotation(title = "Before")
# Sample distribution after integration. Select the reduction parameter below according to the integration method used previously
p2 <- DimPlot(integrated, split.by = "Sample" , cols = my36colors, reduction = "atacharmonyumap" ,group.by = "atacneighbor_res.0.5")+plot_annotation(title = "After")
#p2 <- DimPlot(integrated, split.by = "Sample" , cols = my36colors, reduction = "atacintegratedumap",group.by = "atacneighbor_res.0.5")+plot_annotation(title = "After")
p1
p2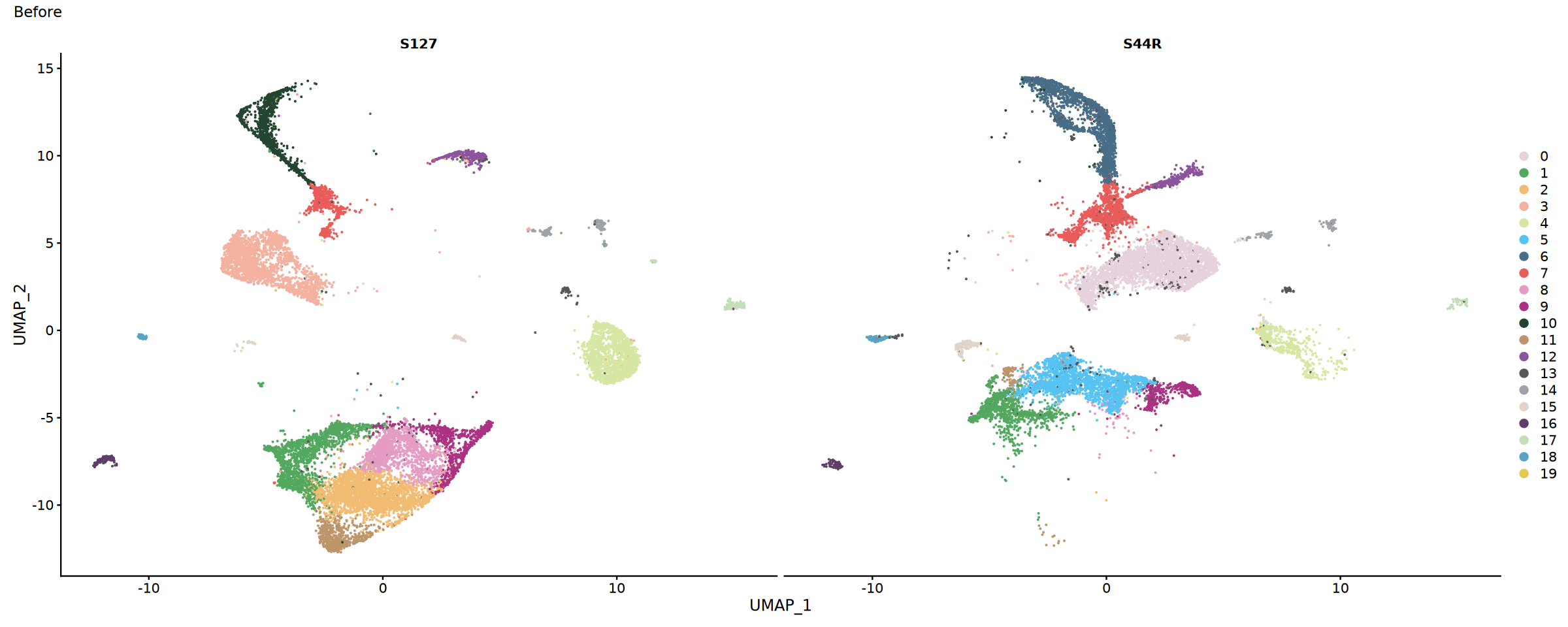
Cell Annotation
Cell type annotation based on gene activity scores, identifying different cell types through known marker genes. Gene activity can only roughly classify scATAC-seq data. Specific annotation strategies for scATAC-seq data include:
(1) Rough classification using gene activity;
(2) Using annotated scRNA-seq of the same tissue as a reference to transfer scRNA-seq cell labels to scATAC-seq to complete cell annotation;
(3) Using CoveragePlot in the Signac package to visualize the specific opening of marker gene peaks to complete cell annotation. This tutorial only demonstrates cell annotation using marker gene activity.
Gene Activity Analysis
Principle: Count peaks signals associated with each gene (e.g., gene body + promoter region within a certain range upstream/downstream) and accumulate them to obtain the "activity score" of that gene in each cell.
- Typical practice takes ~2kb upstream and ~1kb downstream of TSS (adjustable)
- The resulting
ACTIVITYassay will be used to build anchors withRNAassay
# Calculate gene activity score
gene.activities <- GeneActivity(integrated)
# Add gene activity matrix as a new assay
integrated[['RNA']] <- CreateAssayObject(counts = gene.activities)
DefaultAssay(integrated)="RNA"
# Normalize gene activity data
integrated <- NormalizeData(
object = integrated,
assay = 'RNA',
normalization.method = 'LogNormalize',
scale.factor = median(integrated$nCount_RNA)
)Extracting reads overlapping genomic regions
Extracting reads overlapping genomic regions
Collect Typical Marker Genes for Cell Types
Collect marker gene sets for different cell types based on tissue type. This example data is from the eye. Below are the cell types in the eye and their corresponding marker genes. Use a dot plot to visualize which cell types' marker genes are highly open in different clusters.
eye_marker_integrated <- list(
# ========== Photoreceptor Cells ==========
"Rod_Photoreceptors" = c("RHO", "PDE6B", "CNGB1", "PDE6A", "NR2E3", "REEP6",
"RCVRN", "SAG", "NEB", "SLC24A3", "TRPM1"),
"Cone_Photoreceptors" = c("ARR3", "GNAT2", "OPN1SW", "OPN1MW", "TRPM3"),
# ========== Retinal Neurons ==========
"Bipolar_Cells" = c("VSX1", "VSX2", "OTX2", "GRM6", "PRKCA",
"DLG2", "NRXN3", "RBFOX3", "FSTL4"),
"Amacrine_Cells" = c("GAD1", "GAD2", "C1QL2", "TFAP2B", "SLC32A1"),
"Horizontal_Cells" = c("ONECUT1", "LHX1", "CALB1", "NFIA"),
"Retinal_Ganglion_Cells" = c("RBPMS", "THY1", "NEFL", "POU4F2"),
# ========== Glial Cells ==========
"Muller_Glia" = c("SLC1A3", "RLBP1", "SOX9", "CRYAB"),
"Astrocytes" = c("GFAP", "AQP4", "S100B"),
"Microglia" = c("TMEM119", "C1QA", "AIF1", "CX3CR1", "CD74",
"PTPRC", "CSF1R", "CCL3L1", "SPP1", "P2RY12"),
# ========== Vascular and Support Cells ==========
"Endothelial" = c("FLT1", "PODXL", "PLVAP", "KDR", "EGFL7",
"CLDN5", "PECAM1", "CDH5"),
"Pericytes" = c("PDGFRB", "RGS5", "CSPG4", "STAB1"),
# ========== Epithelial Cells ==========
"RPE" = c("RPE65", "BEST1", "PMEL", "TYRP1", "DCT", "SLC38A11"),
"Corneal_Epithelial" = c("KRT12", "KRT3", "PAX6"),
"Corneal_Endothelial" = c("SLC4A11", "COL8A2", "ATP1A1"),
"Conjunctival_Epithelial" = c("KRT13", "KRT19", "MUC5AC"),
# ========== Lens Cells ==========
"Lens_Epithelial" = c("CRYAA", "BFSP1", "MIP"),
"Lens_Fiber" = c("CRYGS", "LIM2", "FN1"),
# ========== Other Cells ==========
"Melanocytes" = c("TYR", "MLANA", "MITF"),
"Erythrocytes" = c("HBB", "HBA1", "HBA2"),
"ECM" = c("COL1A1", "COL3A1", "COL12A1", "COL1A2", "COL6A3"),
"Others" = c("TTN", "CLCN5", "DCC", "MIAT")
)# Set plot size
options(repr.plot.width=23, repr.plot.height=7)
# Draw DotPlot
DotPlot(integrated,
group.by = "atacneighbor_res.0.5",
features = eye_marker_integrated,
cols = c("#f8f8f8","#ff3472"),
dot.min = 0.05,
dot.scale = 8)+ # Apply custom color scheme
RotatedAxis() +
scale_x_discrete("") +
scale_y_discrete("") +
theme(
axis.text.x = element_text(size = 12, face = "bold",
angle = 45, hjust = 1, vjust = 1),
axis.text.y = element_text(size = 12, face = "bold"),
plot.title = element_text(size = 14, face = "bold", hjust = 0.5),
legend.title = element_text(size = 10, face = "bold")
) +
ggtitle("Marker Genes Expression") +
labs(color = "Expression\nLevel") # Modify legend title"The following requested variables were not found: SLC24A3, TRPM3, PRKCA, DLG2, NRXN3, NFIA, CCL3L1, DCC, MIAT"
"Removed 530 rows containing missing values or values outside the scale range
(\`geom_point()\`)."
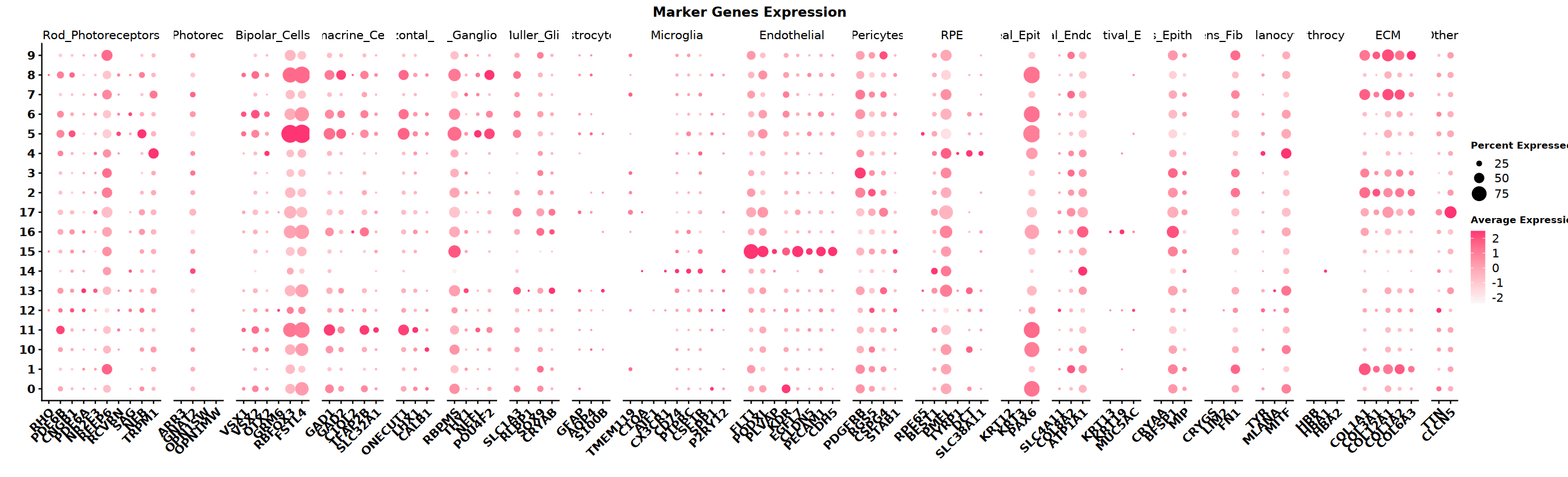
Cell Type Labeling
Assign cell type labels to each cluster based on marker gene opening patterns (i.e., gene activity status).
# Define cell types based on clustering results and marker gene expression
# Note: The specific mapping from clusters to cell types needs to be adjusted based on marker gene expression results
integrated$celltype <- recode(integrated$atacneighbor_res.0.5,
"0" = "Photoreceptors",
"1" = "ECM",
"2" = "ECM",
"3" = "ECM",
"4" = "Rod_photoreceptors",
"5" = "Bipolar_Cells",
"6" = "Amacrine_Horizontal",
"7" = "ECM",
"8" = "Bipolar_Cells",
"9" = "ECM",
"10" = "Photoreceptors",
"11" = "Amacrine_Horizontal",
"12" = "Photoreceptors",
"13" = "Muller_Glia",
"14" = "Microglia",
"15" = "Endothelial",
"16" = "Muller_Glia",
"17" = "ECM"
)# Basic UMAP plot
p1 <- DimPlot(integrated,
cols = my36colors,
group.by = "celltype",
label = TRUE) +
ggtitle("Cell Type Annotation")
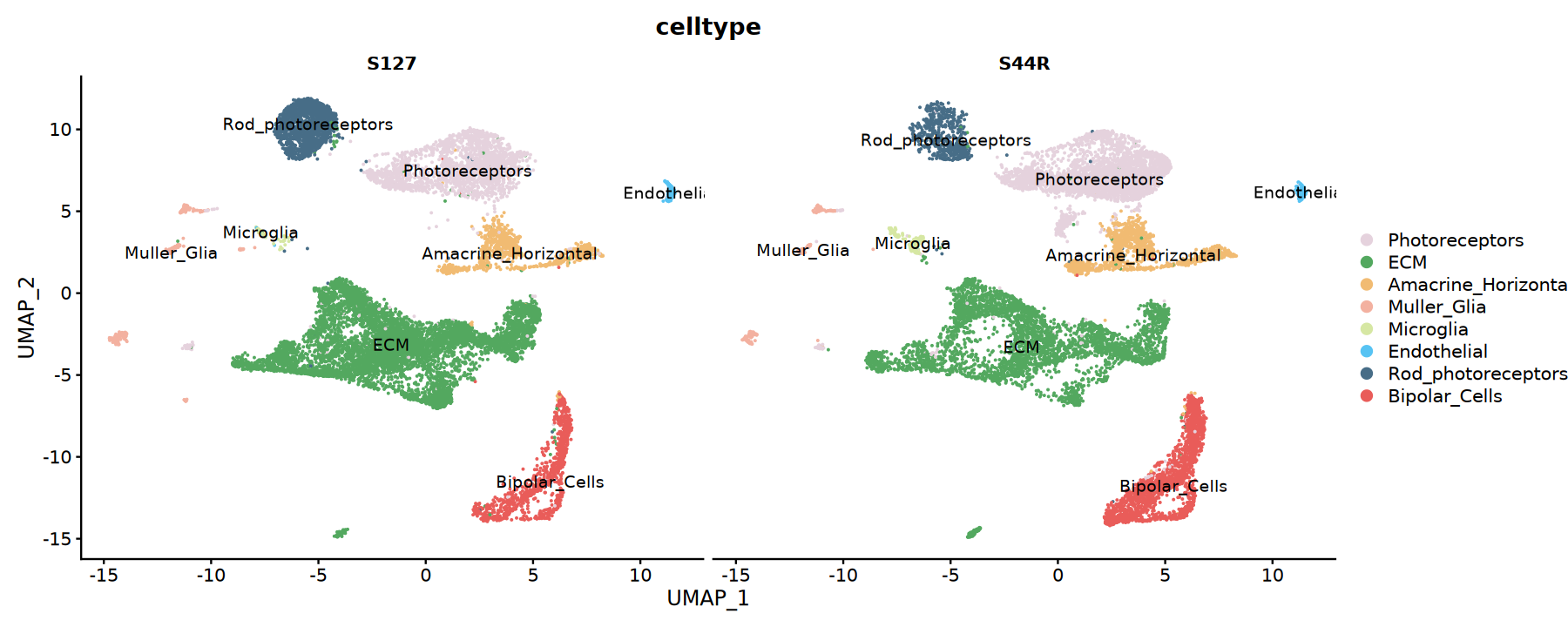
# Split sample UMAP plot
p2 <- DimPlot(integrated,
cols = my36colors,
split.by = "Sample",
group.by = "celltype",
label = TRUE)
# Save PDF (high quality vector graphic)
ggsave('./celltype_annotation_umap.pdf', p1, width = 8, height = 6)
ggsave('./celltype_by_sample_umap.pdf', p2, width = 16, height = 6)
# Display graphics
# Set plot dimensions
options(repr.plot.width=9, repr.plot.height=6)
print(p1)
# Set plot dimensions
options(repr.plot.width=15, repr.plot.height=6)
print(p2)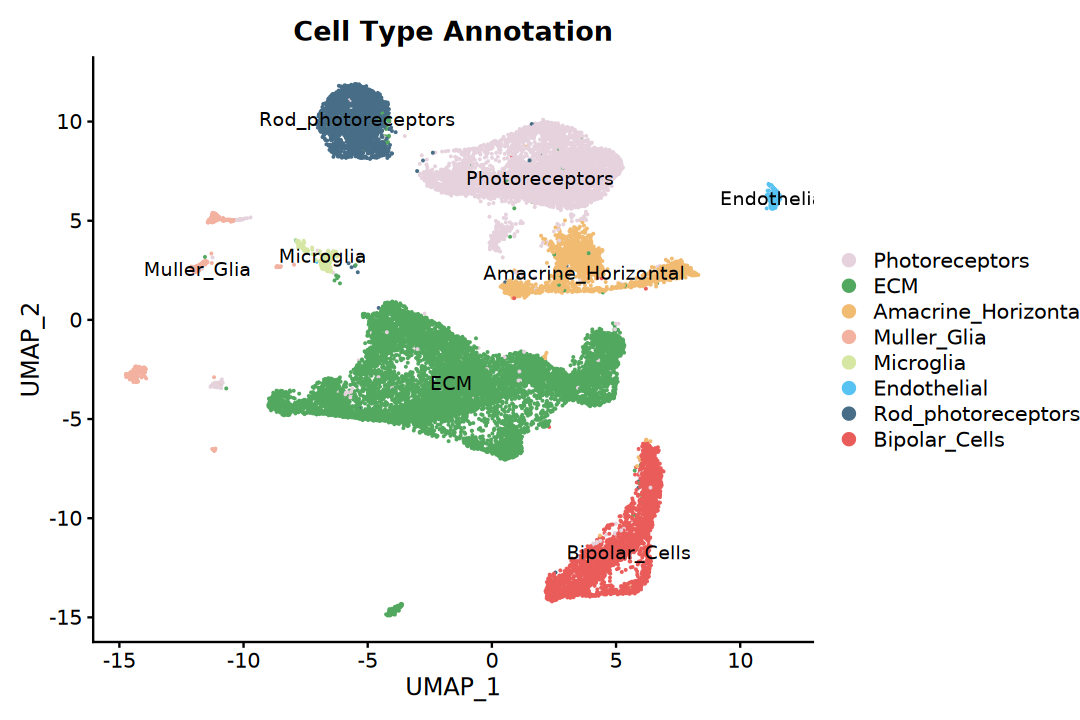
Save Results
Save the integrated object and key charts. It is recommended to save intermediate objects at the same time for reproduction and subsequent review.
saveRDS(integrated, file = "./integrated.rds")Summary
This tutorial demonstrates the complete workflow for scATAC-seq multi-sample integration analysis:
Key Technical Points
- TF-IDF Normalization: Suitable for sparse scATAC-seq data
- LSI Dimensionality Reduction: Latent Semantic Indexing, more suitable for scATAC-seq data than PCA
- Peaks Merging: Unifying peak sets from different samples is a key step in integration
- Gene Activity Score: Convert peaks signals to gene-level activity scores
Notes
- QC Thresholds: Filtering parameters need to be adjusted based on specific data characteristics
- Integration Parameters: LSI dimension selection and integration parameters need optimization
- Cell Annotation: Manual annotation requires combining biological knowledge and marker gene expression
- Computational Resources: scATAC-seq data volume is large and requires sufficient memory and computational resources
Suggestions for Subsequent Analysis
- Differential Accessibility Region (DAR) Analysis
- Transcription Factor Footprint Analysis
- Gene Regulatory Network Inference
- Multi-omics Integration Analysis with scRNA-seq Data
- Cell Trajectory and Pseudotime Analysis
sessionInfo()Platform: x86_64-conda-linux-gnu (64-bit)
Running under: Debian GNU/Linux 12 (bookworm)
Matrix products: default
BLAS/LAPACK: /jp_envs/envs/common/lib/libopenblasp-r0.3.29.so; LAPACK version 3.12.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
time zone: Asia/Shanghai
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] future_1.40.0 patchwork_1.3.0
[3] ggplot2_3.5.2 dplyr_1.1.4
[5] biovizBase_1.50.0 BSgenome.Hsapiens.UCSC.hg38_1.4.5
[7] BSgenome_1.70.1 rtracklayer_1.62.0
[9] BiocIO_1.12.0 Biostrings_2.70.1
[11] XVector_0.42.0 EnsDb.Hsapiens.v86_2.99.0
[13] ensembldb_2.26.0 AnnotationFilter_1.26.0
[15] GenomicFeatures_1.54.1 AnnotationDbi_1.64.1
[17] Biobase_2.62.0 GenomicRanges_1.54.1
[19] GenomeInfoDb_1.38.1 IRanges_2.36.0
[21] S4Vectors_0.40.2 BiocGenerics_0.48.1
[23] Signac_1.10.0 SeuratObject_4.1.4
[25] Seurat_4.4.0 repr_1.1.7
loaded via a namespace (and not attached):
[1] ProtGenerics_1.34.0 matrixStats_1.5.0
[3] spatstat.sparse_3.1-0 bitops_1.0-9
[5] httr_1.4.7 RColorBrewer_1.1-3
[7] tools_4.3.3 sctransform_0.4.1
[9] backports_1.5.0 R6_2.6.1
[11] lazyeval_0.2.2 uwot_0.2.3
[13] withr_3.0.2 sp_2.2-0
[15] prettyunits_1.2.0 gridExtra_2.3
[17] progressr_0.15.1 cli_3.6.4
[19] Cairo_1.6-2 spatstat.explore_3.4-2
[21] labeling_0.4.3 spatstat.data_3.1-6
[23] ggridges_0.5.6 pbapply_1.7-2
[25] Rsamtools_2.18.0 pbdZMQ_0.3-13
[27] foreign_0.8-87 R.utils_2.13.0
[29] dichromat_2.0-0.1 parallelly_1.43.0
[31] rstudioapi_0.15.0 RSQLite_2.3.9
[33] generics_0.1.3 ica_1.0-3
[35] spatstat.random_3.3-3 Matrix_1.6-5
[37] ggbeeswarm_0.7.2 abind_1.4-5
[39] R.methodsS3_1.8.2 lifecycle_1.0.4
[41] yaml_2.3.10 SummarizedExperiment_1.32.0
[43] SparseArray_1.2.2 BiocFileCache_2.10.1
[45] Rtsne_0.17 grid_4.3.3
[47] blob_1.2.4 promises_1.3.2
[49] crayon_1.5.3 miniUI_0.1.1.1
[51] lattice_0.22-7 cowplot_1.1.3
[53] KEGGREST_1.42.0 pillar_1.10.2
[55] knitr_1.49 rjson_0.2.23
[57] future.apply_1.11.3 codetools_0.2-20
[59] fastmatch_1.1-6 leiden_0.4.3.1
[61] glue_1.8.0 spatstat.univar_3.1-2
[63] data.table_1.17.0 vctrs_0.6.5
[65] png_0.1-8 gtable_0.3.6
[67] cachem_1.1.0 xfun_0.50
[69] S4Arrays_1.2.0 mime_0.13
[71] survival_3.8-3 RcppRoll_0.3.1
[73] fitdistrplus_1.2-2 ROCR_1.0-11
[75] nlme_3.1-168 bit64_4.5.2
[77] progress_1.2.3 filelock_1.0.3
[79] RcppAnnoy_0.0.22 irlba_2.3.5.1
[81] vipor_0.4.7 KernSmooth_2.23-26
[83] rpart_4.1.23 colorspace_2.1-1
[85] DBI_1.2.3 Hmisc_5.2-1
[87] nnet_7.3-19 ggrastr_1.0.2
[89] tidyselect_1.2.1 bit_4.5.0.1
[91] compiler_4.3.3 curl_6.0.1
[93] htmlTable_2.4.3 xml2_1.3.6
[95] DelayedArray_0.28.0 plotly_4.10.4
[97] checkmate_2.3.2 scales_1.3.0
[99] lmtest_0.9-40 rappdirs_0.3.3
[101] stringr_1.5.1 digest_0.6.37
[103] goftest_1.2-3 spatstat.utils_3.1-3
[105] rmarkdown_2.29 htmltools_0.5.8.1
[107] pkgconfig_2.0.3 base64enc_0.1-3
[109] MatrixGenerics_1.14.0 dbplyr_2.5.0
[111] fastmap_1.2.0 rlang_1.1.5
[113] htmlwidgets_1.6.4 shiny_1.10.0
[115] farver_2.1.2 zoo_1.8-14
[117] jsonlite_2.0.0 BiocParallel_1.36.0
[119] R.oo_1.27.0 VariantAnnotation_1.48.1
[121] RCurl_1.98-1.16 magrittr_2.0.3
[123] Formula_1.2-5 GenomeInfoDbData_1.2.11
[125] IRkernel_1.3.2 munsell_0.5.1
[127] Rcpp_1.0.14 reticulate_1.42.0
[129] stringi_1.8.7 zlibbioc_1.48.0
[131] MASS_7.3-60.0.1 plyr_1.8.9
[133] parallel_4.3.3 listenv_0.9.1
[135] ggrepel_0.9.6 deldir_2.0-4
[137] IRdisplay_1.1 splines_4.3.3
[139] tensor_1.5 hms_1.1.3
[141] igraph_2.0.3 uuid_1.2-1
[143] spatstat.geom_3.3-6 reshape2_1.4.4
[145] biomaRt_2.58.0 XML_3.99-0.17
[147] evaluate_1.0.3 httpuv_1.6.15
[149] RANN_2.6.2 tidyr_1.3.1
[151] purrr_1.0.4 polyclip_1.10-7
[153] scattermore_1.2 xtable_1.8-4
[155] restfulr_0.0.15 later_1.4.2
[157] viridisLite_0.4.2 tibble_3.2.1
[159] memoise_2.0.1 beeswarm_0.4.0
[161] GenomicAlignments_1.38.0 cluster_2.1.8.1
[163] globals_0.16.3