scFAST-seq Mutation Analysis: Data Merging, UMAP Display and Integration
Select R environment in the top right corner
library(Seurat)
library(dplyr)
library(Matrix)
library(stringr)
library(ggplot2)
options(repr.plot.width = 12, repr.plot.height = 6)will retire shortly. Please refer to R-spatial evolution reports on
https://r-spatial.org/r/2023/05/15/evolution4.html for details.
This package is now running under evolution status 0
rgeos version: 0.6-3, (SVN revision 696)
GEOS runtime version: 3.11.2-CAPI-1.17.2
Please note that rgeos will be retired during October 2023,
plan transition to sf or terra functions using GEOS at your earliest convenience.
See https://r-spatial.org/r/2023/05/15/evolution4.html for details.
GEOS using OverlayNG
Linking to sp version: 1.6-0
Polygon checking: TRUE
Attaching SeuratObject
Attaching sp
Attaching package: ‘dplyr’
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
Merge mutation data into single-sample rds
Input Data
- Single-sample rds (can select rds corresponding to analysis workflow)
- Two mutation matrices for this sample (all/alt, contact account manager to release, limited to full-length sequence data)
Matrix files can be uploaded by clicking upload in the top left corner

Content to Modify
Modify rds path below, relative directory is ../data/, absolute directory is /home/mambauser/data/
Modify mutation matrix filenames below
#load rds and matrix
obj = readRDS("data/AY1732591902625/input.rds") # AY1732591902625 is workflow ID
# AY1732591902625 is workflow ID
meta = read.table("data/AY1732591902625/meta.tsv", header = T, sep = "\t", check.names = F)
rownames(meta) = meta$barcode
obj = AddMetaData(object = obj,metadata = meta)
snv_cover_mat = read.delim("PBMC.snp_indel.all_UMI.matrix", header = T, row.names = 1)
snv_mut_mat = read.delim("PBMC.snp_indel.alt_UMI.matrix", header = T, row.names = 1)View Matrix
snv_cover_mat[1:3, 1:3]
snv_mut_mat[1:3, 1:3]| AAGTTCGTACTGGTTCT | CTGCAGGTACGGAGTAG | TAACGACCGACTGCGCA | |
|---|---|---|---|
| <int> | <int> | <int> | |
| SDF4:chr1-1223263:T>G | 0 | 0 | 0 |
| SLC35E2B:chr1-1668373:C>T | 0 | 0 | 0 |
| CDK11A:chr1-1709071:C>T | 0 | 0 | 0 |
| AAGTTCGTACTGGTTCT | CTGCAGGTACGGAGTAG | TAACGACCGACTGCGCA | |
|---|---|---|---|
| <int> | <int> | <int> | |
| SDF4:chr1-1223263:T>G | 0 | 0 | 0 |
| SLC35E2B:chr1-1668373:C>T | 0 | 0 | 0 |
| CDK11A:chr1-1709071:C>T | 0 | 0 | 0 |
# Function:process cover/mut matrix
## object : Seurat object
## mat : matrix or dgCMatrix or data.frame
## assay_name: specified new assay name for matrix
generate_mat = function(object, mat, assay_name) {
#make sure consistent of barcodes
diff_barcode = setdiff(colnames(object), colnames(mat))
if (length(diff_barcode) == ncol(object)) {
stop("no common barcodes between matrix and seurat object")
}
if (length(diff_barcode) != 0) {
diff_matrix = matrix(0,
nrow = nrow(mat),
ncol = length(diff_barcode),
dimnames = list(rownames(mat), diff_barcode))
mat = cbind(mat, diff_matrix)
}
mat = as(as.matrix(mat), 'dgCMatrix')
#reorder barcode according rds
mat = mat[, colnames(object)]
#add new assay into rds
object[[assay_name]] = CreateAssayObject(mat)
return(object)
}Run this command to add mutation matrix and coverage matrix to obj respectively
#cover matrix
obj = generate_mat(object = obj, mat = snv_cover_mat, assay_name = "SNV_all")
#mutation matrix
obj = generate_mat(object = obj, mat = snv_mut_mat, assay_name = "SNV")“Keys should be one or more alphanumeric characters followed by an underscore, setting key from snv_all_ to snvall_”
obj has three assays, added successfully
#result
Assays(obj)- 'RNA'
- 'SNV_all'
- 'SNV'
#check value
obj@assays$SNV[1:3,1:3]AAGTTCGTACTGGTTCT CTGCAGGTACGGAGTAG CCTCTAGATGTAATTCC
SDF4:chr1-1223263:T>G . . .
SLC35E2B:chr1-1668373:C>T . . .
CDK11A:chr1-1709071:C>T . . .
Save rds
saveRDS(obj, file = "add_snv.rds")Display Mutation Occurrence
Display Single Mutation Occurrence in Cell Population
Specify assay as SNV, i.e., specify mutation matrix, display cells with this mutation
Example mutation: SLC35E2B:chr1-1668373:C>T
DefaultAssay(obj) = "SNV"
FeaturePlot(obj, features = "SLC35E2B:chr1-1668373:C>T",ncol = 2) +
DimPlot(obj, group.by = "resolution.0.8", label = T)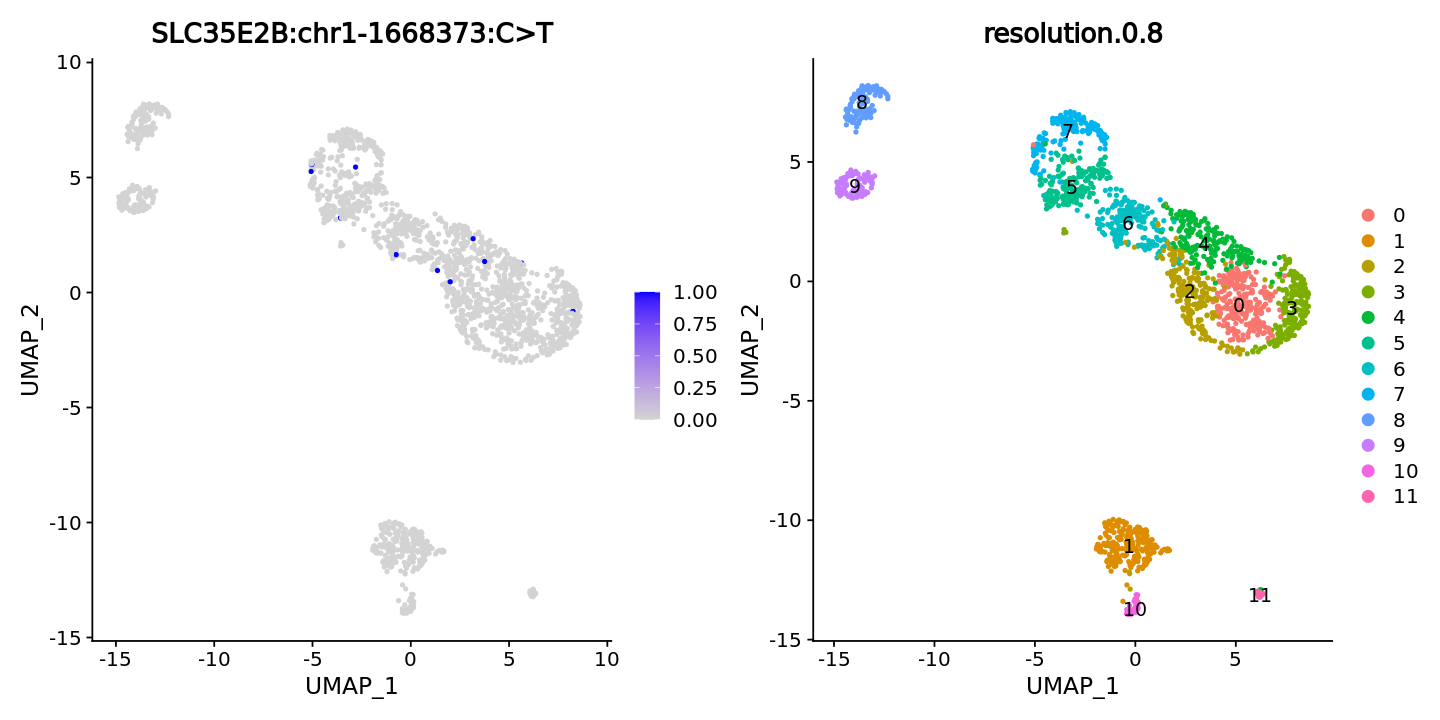
Save Image
Image filename is SLC35E2B_C_to_T_mut.png, saved in current directory, can be downloaded from the left
p = FeaturePlot(obj, features = "SLC35E2B:chr1-1668373:C>T",ncol = 2) +
DimPlot(obj, group.by = "resolution.0.8", label = T)
ggsave(p, file = "SLC35E2B_C_to_T_mut.png", width = 12, height = 6)Display Multiple Mutations Occurrence in Cell Population
Specify assay as SNV, i.e., specify mutation matrix, display cells with this mutation
Example specifying top 5 mutations
DefaultAssay(obj) = "SNV"
head5_mut = head(rownames(obj), 5)
head5_mut- 'SDF4:chr1-1223263:T>G'
- 'SLC35E2B:chr1-1668373:C>T'
- 'CDK11A:chr1-1709071:C>T'
- 'RPL22:chr1-6197724:C>T'
- 'DNAJC11:chr1-6667742:TTC>-'
Batch Save Images
for ( i in head5_mut) {
p = FeaturePlot(obj, features = i, ncol = 2)
filename = stringr::str_replace_all(i, "[:>]", "_")
ggsave(p, file = paste0(filename, ".png"), width = 6, height = 6) # Saved in current directory
}Display Cell Population Covering This Site
Specify assay as SNV_all, i.e., specify coverage matrix, display cells covering the site of this mutation
DefaultAssay(obj) = "SNV_all"
FeaturePlot(obj, features = "SLC35E2B:chr1-1668373:C>T",ncol = 2) +
DimPlot(obj, group.by = "resolution.0.8", label = T)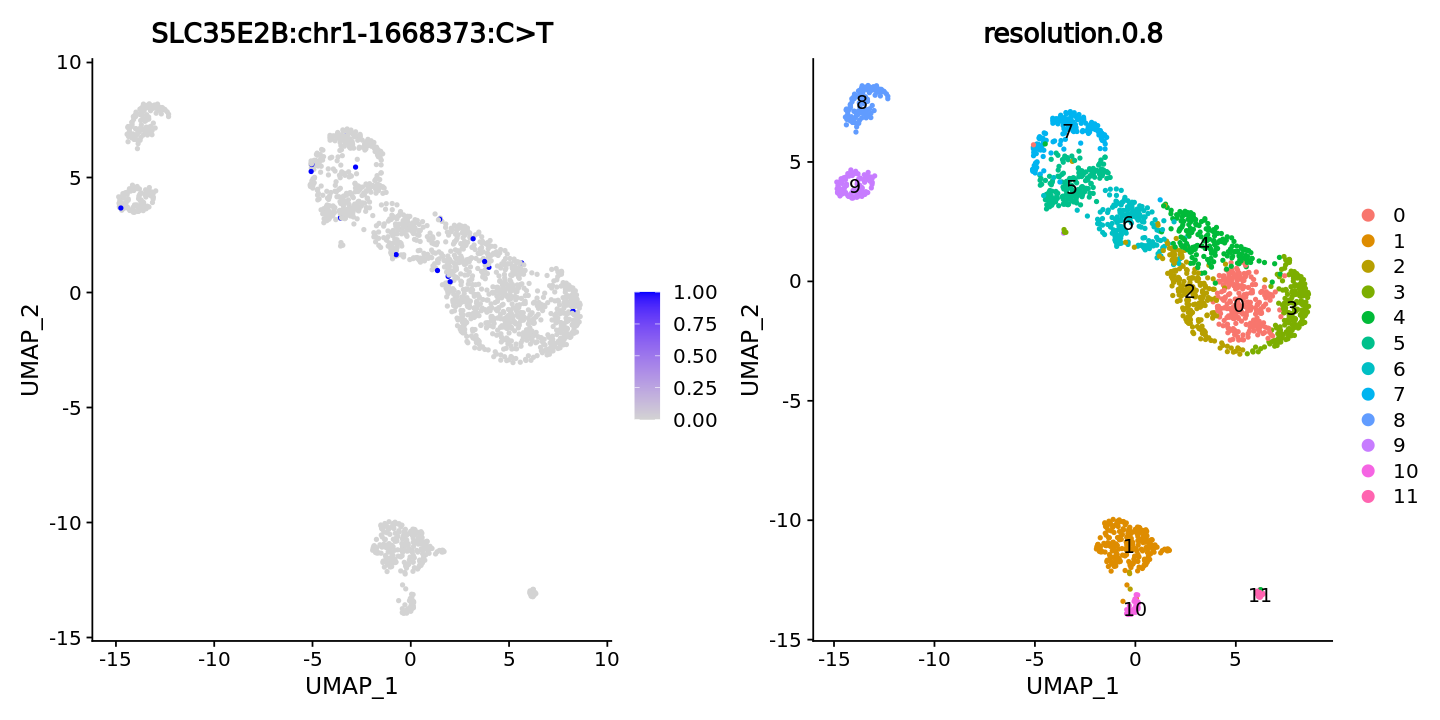
Display Expression of Gene Containing Mutation
Specify assay as RNA, i.e., specify expression data, display expression of the gene containing this mutation
DefaultAssay(obj) = "RNA"
FeaturePlot(obj, features = "SLC35E2B",ncol = 2) +
DimPlot(obj, group.by = "resolution.0.8", label = T)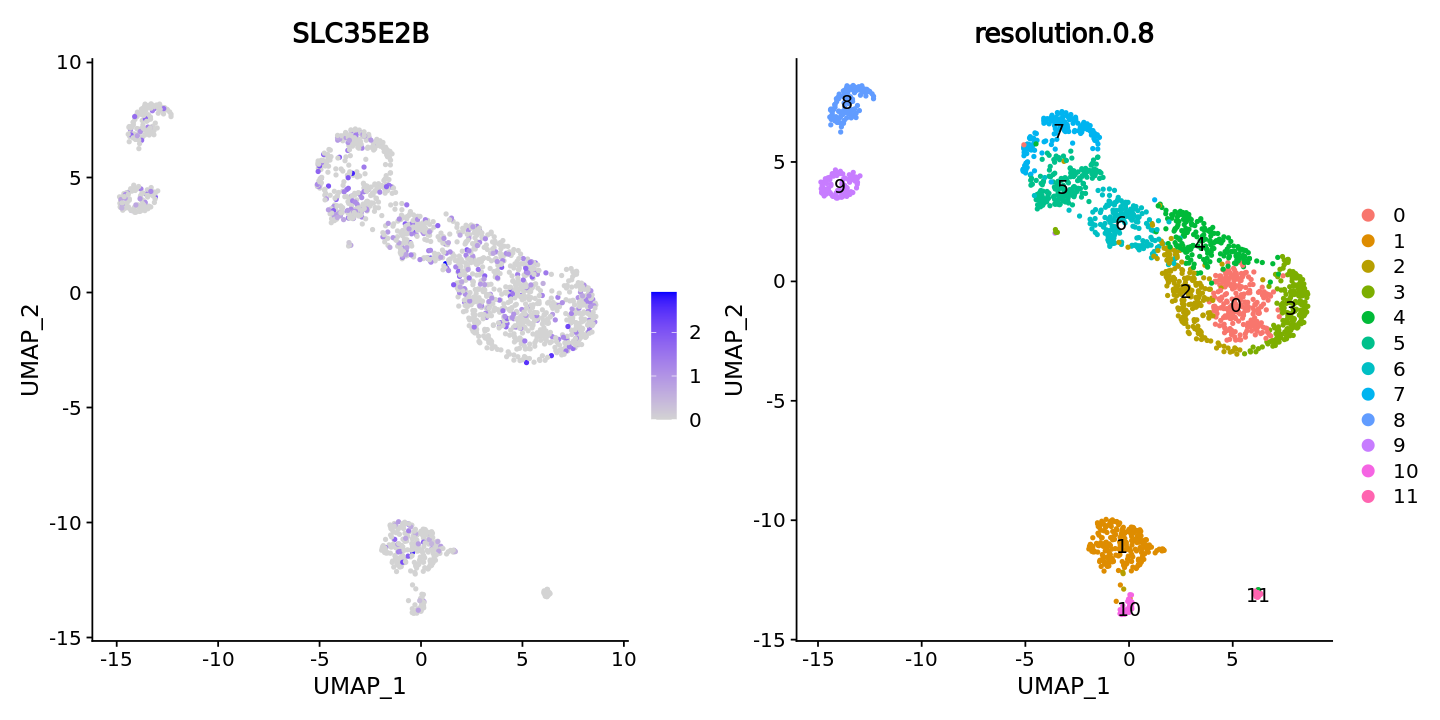
Add Mutation Status as New Label
Classify cells into two categories: Mutated/Not Mutated, and display as a new label;
Example mutation "SLC35E2B:chr1-1668373:C>T"
interest_mut = "SLC35E2B:chr1-1668373:C>T"
DefaultAssay(obj) = "SNV"
mut_label = FetchData(obj, interest_mut)
mut_label = ifelse(mut_label == 0, "negative", "positive")
table(mut_label)negative positive
1909 12
Add Label
obj = AddMetaData(obj, metadata = mut_label, col.name = "SLC35E2B_C_to_T")Display
DefaultAssay(obj) = "RNA"
FeaturePlot(obj, features = "SLC35E2B",ncol = 2) +
DimPlot(obj, group.by = "SLC35E2B_C_to_T", cols = c("grey","red"))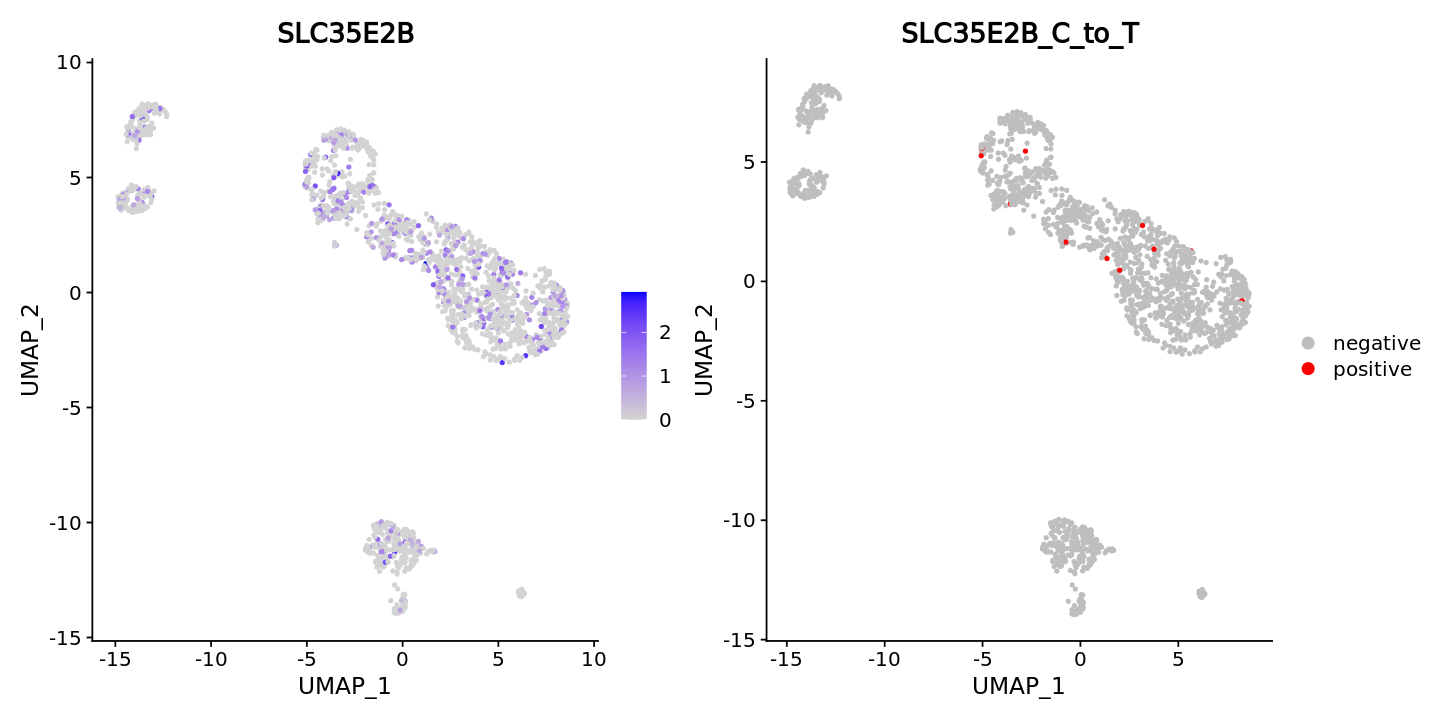
Export Table
df = data.frame(barcode = colnames(obj),
SLC35E2B_C_to_T = mut_label)
head(df)
write.csv(df, file="SLC35E2B_C_to_T_mut_meta.csv", quote=F, row.names = F)| barcode | SLC35E2B.chr1.1668373.C.T | |
|---|---|---|
| <chr> | <chr> | |
| AAGTTCGTACTGGTTCT | AAGTTCGTACTGGTTCT | negative |
| CTGCAGGTACGGAGTAG | CTGCAGGTACGGAGTAG | negative |
| CCTCTAGATGTAATTCC | CCTCTAGATGTAATTCC | negative |
| TAACGACCGACTGCGCA | TAACGACCGACTGCGCA | negative |
| ATCAGGTGCTTTCAGAC | ATCAGGTGCTTTCAGAC | negative |
| GACCCTTTACGTGTTCC | GACCCTTTACGTGTTCC | negative |
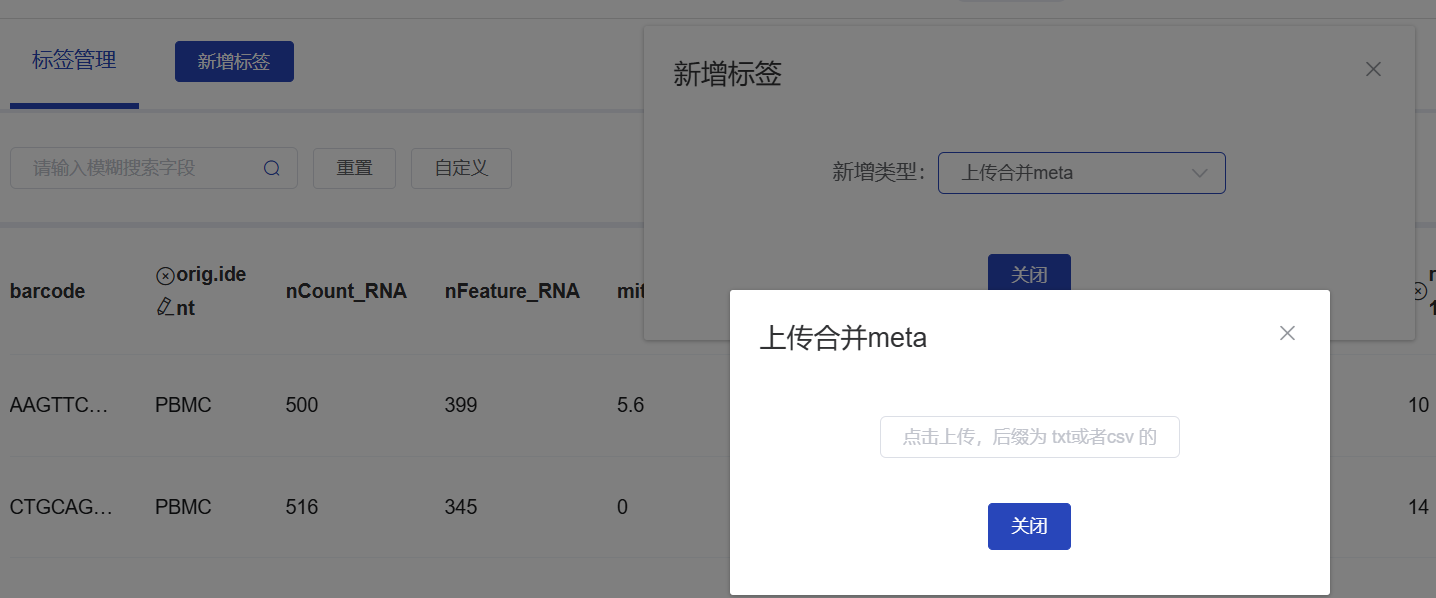
Upload Table to Cloud Platform
Table saved to current path, click download on the left, enter cloud platform
Cloud Platform - Select Analysis Workflow - Auxiliary Info - Label Management - Add Label - Upload Merged Meta - Click Upload
Multi-Sample Integration of Full-Length Mutation Data
Merge mutation matrices of multiple samples with integrated rds, enabling mutation correlation analysis across samples.
Input Data
- Integrated rds. (Can select corresponding analysis workflow)
- Mutation occurrence matrix and coverage matrix for each sample (all/alt, contact account manager to release, limited to full-length sequence data)
Example: SAMple column in integrated rds is "PBMC", mutation matrix filename is "PBMC.snp_indel.all_UMI.matrix", "PBMC.snp_indel.alt_UMI.matrix"
Matrix files can be uploaded by clicking upload in the top left corner
Content to Modify
Modify rds path below, relative directory is ../data/, absolute directory is /home/mambauser/data/
Modify mutation matrix filenames below
##read integrated rds
integrate_obj = readRDS("integrated_seurat.rds") # or integrate_obj = readRDS("data/WorkflowID/input.rds")
unique(integrate_obj$Sample)
# Matrix files for snv_cover_mat & snv_mut_mat must be in the current working directory, you can change working directory via setwd()
snv_cover_mat = c("PBMC.snp_indel.all_UMI.matrix", "cellline.snp_indel.all_UMI.matrix")
snv_mut_mat = c("PBMC.snp_indel.alt_UMI.matrix", "cellline.snp_indel.alt_UMI.matrix")- 'PBMC'
- 'cellline'
# Function:process cover/mut matrix
## object : Seurat object
## mat : matrix or dgCMatrix or data.frame
## assay_name: specified new assay name for matrix
generate_mat = function(object, mat, assay_name) {
#make sure consistent of barcodes
diff_barcode = setdiff(colnames(object), colnames(mat))
if (length(diff_barcode) == ncol(object)) {
stop("no common barcodes between matrix and seurat object")
}
if (length(diff_barcode) != 0) {
diff_matrix = matrix(0,
nrow = nrow(mat),
ncol = length(diff_barcode),
dimnames = list(rownames(mat), diff_barcode))
mat = cbind(mat, diff_matrix)
}
mat = as(as.matrix(mat), 'dgCMatrix')
#reorder barcode according rds
mat = mat[, colnames(object)]
#add new assay into rds
object[[assay_name]] = CreateAssayObject(mat)
return(object)
}
# Function:process cover/mut matrix for integrated rds
## object: Seurat object
## snv_cover_mat: vector, cover matrix for each sample
## snv_mut_mat:vector, mut matrix for each sample
## meta_name: column name in meta.data, contains samples' name, same as marix filename's prefix
## cover_assay_name : new assay name for cover matrix
## mut_assay_name : new assay name for mut matrix
generate_mat_integrated = function(object,
snv_cover_mat,
snv_mut_mat,
meta_name = "Sample",
cover_assay_name = "SNV_all",
mut_assay_name = "SNV") {
#get barcode suffix for each sample
suffix = as.data.frame(object@meta.data) %>%
select(sym(meta_name)) %>%
unique()
suffix$barcode = rownames(suffix)
suffix$suffix = stringr::str_replace_all(suffix$barcode,
"[A-Z]{17}", "")
#check sample name
cover_sample = stringr::str_replace_all(snv_cover_mat,
".snp_indel.all_UMI.matrix","")
mut_sample = stringr::str_replace_all(snv_mut_mat,
".snp_indel.alt_UMI.matrix","")
if(! identical(cover_sample, mut_sample)) {
stop("filenames are not same for snv_cover_mat and snv_mut_mat")
}
if(! all(suffix[[meta_name]] %in% cover_sample)) {
stop("some samples do not have corresponding matrix files")
}
#get all matrix, add suffix
cover_mat_list = lapply(1:nrow(suffix), function(e) {
mat = read.delim(paste0(suffix[[meta_name]][e], ".snp_indel.all_UMI.matrix"),
header = T, row.names = 1)
colnames(mat) = paste0(colnames(mat), suffix[["suffix"]][e])
as.matrix(mat)
})
mut_mat_list = lapply(1:nrow(suffix), function(e) {
mat = read.delim(paste0(suffix[[meta_name]][e], ".snp_indel.alt_UMI.matrix"),
header = T, row.names = 1)
colnames(mat) = paste0(colnames(mat), suffix[["suffix"]][e])
as.matrix(mat)
})
##define two matrix
all_muts = unique(unlist(lapply(cover_mat_list, rownames)))
all_barcodes = unlist(lapply(cover_mat_list, colnames))
all_cover_mat = Matrix::Matrix(0,
nrow = length(all_muts),
ncol = length(all_barcodes),
dimnames = list(all_muts, all_barcodes))
all_mut_mat = Matrix::Matrix(0,
nrow = length(all_muts),
ncol = length(all_barcodes),
dimnames = list(all_muts, all_barcodes))
#fill in value
for (i in seq.int(cover_mat_list)) {
mat = cover_mat_list[[i]]
all_cover_mat[rownames(mat), colnames(mat)] = mat
mat = mut_mat_list[[i]]
all_mut_mat[rownames(mat), colnames(mat)] = mat
}
#add new assay, function is from top-line in this file
object = generate_mat(object,
mat = all_cover_mat,
assay_name = cover_assay_name)
object = generate_mat(object,
mat = all_mut_mat,
assay_name = mut_assay_name)
}Run command to merge data
integrate_obj = generate_mat_integrated(integrate_obj,
snv_cover_mat,
snv_mut_mat,
meta_name = "Sample")“Keys should be one or more alphanumeric characters followed by an underscore, setting key from snv_all_ to snvall_”
New object has three assays, added successfully
Assays(integrate_obj)- 'RNA'
- 'SNV_all'
- 'SNV'
Save Data
saveRDS(integrate_obj, file = "integrate_add_snv.rds")Can also merge data first, then integrate
#PBMC data
obj1 = readRDS("PBMC_seurat.rds")
snv_cover_mat1 = read.delim("PBMC.snp_indel.all_UMI.matrix", header = T, row.names = 1)
snv_mut_mat1 = read.delim("PBMC.snp_indel.alt_UMI.matrix", header = T, row.names = 1)
#cover matrix
obj1 = generate_mat(object = obj1, mat = snv_cover_mat1, assay_name = "SNV_all")
#mutation matrix
obj1 = generate_mat(object = obj1, mat = snv_mut_mat1, assay_name = "SNV")“Keys should be one or more alphanumeric characters followed by an underscore, setting key from snv_all_ to snvall_”
#cellline data
obj2 = readRDS("cellline_seurat.rds")
snv_cover_mat2 = read.delim("cellline.snp_indel.all_UMI.matrix", header = T, row.names = 1)
snv_mut_mat2 = read.delim("cellline.snp_indel.alt_UMI.matrix", header = T, row.names = 1)
#cover matrix
obj2 = generate_mat(object = obj2, mat = snv_cover_mat2, assay_name = "SNV_all")
#mutation matrix
obj2 = generate_mat(object = obj2, mat = snv_mut_mat2, assay_name = "SNV")“Keys should be one or more alphanumeric characters followed by an underscore, setting key from snv_all_ to snvall_”
integrate_obj = merge(obj1, obj2)“Some cell names are duplicated across objects provided. Renaming to enforce unique cell names.”
New object has three assays, added successfully
Assays(integrate_obj)- 'RNA'
- 'SNV_all'
- 'SNV'
Save Data
saveRDS(integrate_obj, file = "integrate_add_snv.rds")