Single-cell Methylation + RNA Dual-omics Multi-sample Integration Analysis Pipeline (ALLCools)
Load Python Analysis Packages
import os
import re
import glob
from ALLCools.mcds import MCDS
from ALLCools.clustering import tsne, significant_pc_test, log_scale, lsi, binarize_matrix, filter_regions, cluster_enriched_features, ConsensusClustering, Dendrogram, get_pc_centers
from ALLCools.clustering.doublets import MethylScrublet
from ALLCools.plot import *
import scanpy as sc
import scanpy.external as sce
from harmonypy import run_harmony
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
from matplotlib.lines import Line2D
import warnings
import xarray as xr
from ALLCools.clustering import one_vs_rest_dmg
import pybedtools
from scipy import sparsesamples = ["HC10_12","HC14_21"]
var_dim = 'chrom20k'
obs_dim = 'cell'
mc_type = 'CGN'
quant_type = 'hypo-score'- Input Folder Structure
HC10_12
├── HC10_12_exp
│ └── HC10_12
│ └── Analysis
│ └── step3
│ ├── filtered_feature_bc_matrix
│ └── raw_feature_bc_matrix
└── HC10_12_methy
└── step3
├── allcools_generate_datasets
└── split_BAMs
├── filtered_barcode_reads_counts.csv
└── HC10_12_cells.csv⚠️ Important Note
This tutorial applies to the above directory structure. If your file organization is different, please adjust folder paths or modify file addresses in the code accordingly.
⚠️ Important Note
If the reagent type is DD-MET3, you need to read the correspondence.
gex_mc_bc_map = pd.read_csv("/PROJ2/FLOAT/weiqiuxia/project/20241227_methy/script/SeekSoulMethyl/nf/bin/barcodes/DD-M_bUCB3_whitelist.csv",index_col=None)Single-cell RNA Sequencing Data Multi-sample Integration Analysis Pipeline
Data Preprocessing Pipeline
Normalization and Transformation
Count Normalization
Uses total count normalization to eliminate sequencing depth differences between cells, ensuring fair comparison.Variance Stabilizing Transformation
Applies logarithmic transformation (log1p) so that highly expressed genes do not overly dominate analysis results.
Feature Gene Selection
Identifies 2000 highly variable genes via statistical methods; these genes show significant expression differences between cells and are most likely to reflect important biological signals.
Batch Effect Correction
Application of Harmony Algorithm
Uses the advanced Harmony ensemble learning method to elegantly resolve technical variations between multiple samples:
Cell Atlas Construction
Neighborhood Relationship Calculation
Based on the corrected feature space, constructs a k-nearest neighbor graph (k=30) of cells to quantify similarity relationships.
Clustering Analysis
Uses the Leiden algorithm for cell population identification; this algorithm discovers high-quality community structures. Resolution parameter is set to 0.5 for moderate clustering granularity.
Visualization
UMAP Dimensionality Reduction
Preserves data topology, providing an intuitive view of cell distribution.
# Read Expression data and add meta data to RNA and MET data
obj_rna_list = {}
for i in samples:
s_rna_path = os.path.join(f'{i}', f'{i}_exp',f'{i}', 'Analysis', 'step3', 'filtered_feature_bc_matrix')
obj_rna_list[i] = sc.read_10x_mtx(s_rna_path)
#obj_rna_list[i].obs['batch']=f'{i}'
adata_rna=obj_rna_list[samples[0]].concatenate([obj_rna_list[i] for i in samples[1:] ])
adata_rna.obs['Sample'] = adata_rna.obs['batch'].map(
{f'{index}': value for index, value in enumerate(samples)}
)
sc.pp.normalize_total(adata_rna, inplace=True)
sc.pp.log1p(adata_rna)
sc.pp.highly_variable_genes(adata_rna, n_bins=100, n_top_genes=2000)
adata_rna.raw = adata_rna
adata_rna = adata_rna[:, adata_rna.var.highly_variable]
sc.pp.scale(adata_rna, max_value=10)
sc.tl.pca(adata_rna)
ho = run_harmony(adata_rna.obsm['X_pca'],
meta_data=adata_rna.obs,
vars_use='Sample',
random_state=0,
max_iter_harmony=20)
adata_rna.obsm['X_pca_harmony'] = ho.Z_corr.T
reduc_use = 'X_pca_harmony'
sc.pp.neighbors(adata_rna,n_neighbors=30,use_rep = reduc_use)
sc.tl.umap(adata_rna)
sc.tl.tsne(adata_rna,use_rep=reduc_use)
sc.tl.leiden(adata_rna, resolution=0.5)Loading chunk 0-4254/4254
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/anndata/_core/anndata.py:1763: FutureWarning: The AnnData.concatenate method is deprecated in favour of the anndata.concat function. Please use anndata.concat instead.
See the tutorial for concat at: https://anndata.readthedocs.io/en/latest/concatenation.html
warnings.warn(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/preprocessing/_simple.py:843: UserWarning: Received a view of an AnnData. Making a copy.
view_to_actual(adata)
2025-12-19 09:38:10,630 - harmonypy - INFO - Computing initial centroids with sklearn.KMeans...n 2025-12-19 09:38:17,948 - harmonypy - INFO - sklearn.KMeans initialization complete.
2025-12-19 09:38:18,019 - harmonypy - INFO - Iteration 1 of 20
2025-12-19 09:38:21,758 - harmonypy - INFO - Iteration 2 of 20
2025-12-19 09:38:25,497 - harmonypy - INFO - Iteration 3 of 20
2025-12-19 09:38:28,234 - harmonypy - INFO - Iteration 4 of 20
2025-12-19 09:38:29,424 - harmonypy - INFO - Iteration 5 of 20
2025-12-19 09:38:30,781 - harmonypy - INFO - Converged after 5 iterations
RNA Single-modality Cell Annotation
marker_names = ["CD3D","NKG7","CD8A","CD4","CD79A","MS4A1","JCHAIN","LYZ","LILRA4"]
sc.pl.umap(adata_rna,color = marker_names)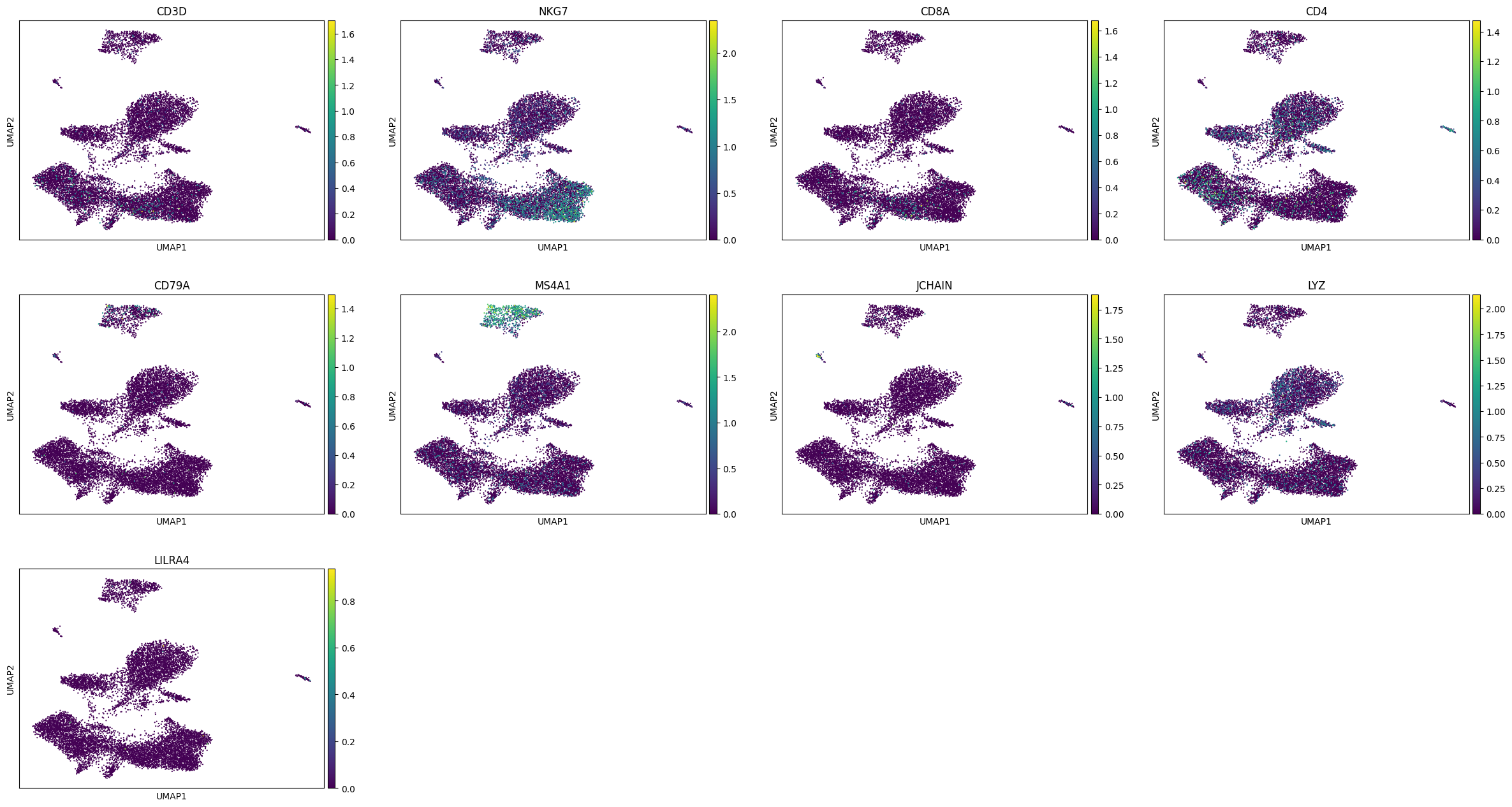
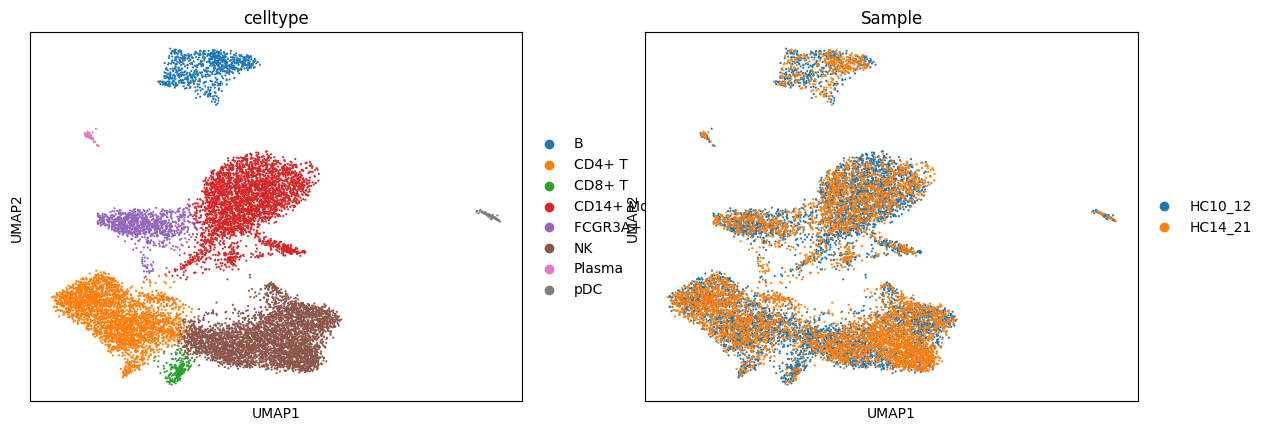
celltype = {
'0': 'NK',
'1': 'CD14+ Mono',
'2': 'CD4+ T',
'3': 'FCGR3A+ Mono',
'4': 'B',
'5': 'CD14+ Mono',
'6': 'CD8+ T',
'7': 'pDC',
'8': 'Plasma'
}
adata_rna.obs['celltype'] = adata_rna.obs['leiden'].map(celltype)sc.pl.umap(adata_rna, color = ['celltype', 'Sample'])cax = scatter(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:394: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
Single-cell Methylation Sequencing Data Analysis Pipeline
Data Preprocessing
Matrix Binarization
Sets 95% threshold for data binarization:
- Above threshold: Marked as hypermethylated state
- Below threshold: Marked as hypomethylated state
- Purpose: To highlight significant epigenetic differences
Region Filtering
Intelligently filters genomic regions, retaining:
- Highly variable methylation regions
- Informative epigenetic markers
Feature Extraction and Dimensionality Reduction
LSI (Latent Semantic Indexing) Analysis
Uses ARPACK algorithm for linear dimensionality reduction, extracting:
- Principal components of genomic methylation patterns
- Epigenetic similarity between cells
- Methylation features in low-dimensional space
Batch Effect Correction
Harmony Ensemble Learning
- Input: LSI reduction results + Sample metadata
- Batch Variable: Sample source (SAMple)
- Random Seed: 0 (Ensures reproducibility)
- Max Iterations: 20 (Balances speed and accuracy)
- Output: Corrected epigenetic space
Correction Effect:
- Cleans technical variation between samples
- Enhances fidelity of biological signals
Cell Atlas Construction
Neighbor Graph Construction
Based on the corrected feature space, builds a 30-nearest neighbor network of cells to quantify epigenetic similarity.
Clustering Analysis
Uses Leiden algorithm for cell population identification:
- Clustering basis: Similarity in hypomethylation features
- Output: Epigenetically defined cell clusters
# MET data integration
# Read Methylation mcds
obj_met_list = {}
mcds_paths = {}
for i in samples:
mcds_path = os.path.join(f'{i}', f'{i}_methy','step3','allcools_generate_datasets', f'{i}.mcds')
mcds_paths[i] = mcds_path
mcds = MCDS.open(mcds_path, obs_dim = 'cell', var_dim = var_dim)
adata = mcds.get_score_adata(mc_type=mc_type, quant_type=quant_type, sparse = True)
obj_met_list[i] = adata
adata_met = obj_met_list[samples[0]].concatenate([obj_met_list[i] for i in samples[1:] ])
adata_met.layers['raw'] = adata_met.X.copy()
adata_met.obs['Sample'] = adata_met.obs['batch'].map(
{f'{index}': value for index, value in enumerate(samples)}
)
binarize_matrix(adata_met, cutoff=0.95)
filter_regions(adata_met)
lsi(adata_met, algorithm='arpack', obsm='X_lsi')
significant_pc_test(adata_met, p_cutoff=0.1, obsm='X_lsi', update=True)
ho = run_harmony(adata_met.obsm['X_lsi'],
meta_data=adata_met.obs,
vars_use='Sample',
random_state=0,
max_iter_harmony=20)
adata_met.obsm['X_lsi_harmony'] = ho.Z_corr.T
reduc_use = 'X_lsi_harmony'
sc.pp.neighbors(adata_met,use_rep = reduc_use, n_neighbors=30)
sc.tl.umap(adata_met)
sc.tl.tsne(adata_met,use_rep=reduc_use)
sc.tl.leiden(adata_met, resolution=0.5)
#adata_met.write_h5ad('adata_met.h5ad')Loading chunk 0-4254/4254
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/anndata/_core/anndata.py:1763: FutureWarning: The AnnData.concatenate method is deprecated in favour of the anndata.concat function. Please use anndata.concat instead.
See the tutorial for concat at: https://anndata.readthedocs.io/en/latest/concatenation.html
warnings.warn(
87948 regions remained.
2025-12-19 09:43:25,691 - harmonypy - INFO - Computing initial centroids with sklearn.KMeans...n
9 components passed P cutoff of 0.1.
Changing adata.obsm['X_pca'] from shape (8523, 100) to (8523, 9)
2025-12-19 09:43:27,280 - harmonypy - INFO - sklearn.KMeans initialization complete.
2025-12-19 09:43:27,315 - harmonypy - INFO - Iteration 1 of 20
2025-12-19 09:43:29,489 - harmonypy - INFO - Iteration 2 of 20
2025-12-19 09:43:31,702 - harmonypy - INFO - Iteration 3 of 20
2025-12-19 09:43:33,883 - harmonypy - INFO - Iteration 4 of 20
2025-12-19 09:43:36,109 - harmonypy - INFO - Iteration 5 of 20
2025-12-19 09:43:38,285 - harmonypy - INFO - Iteration 6 of 20
2025-12-19 09:43:40,491 - harmonypy - INFO - Iteration 7 of 20
2025-12-19 09:43:42,737 - harmonypy - INFO - Iteration 8 of 20
2025-12-19 09:43:44,954 - harmonypy - INFO - Iteration 9 of 20
2025-12-19 09:43:46,153 - harmonypy - INFO - Iteration 10 of 20
2025-12-19 09:43:47,429 - harmonypy - INFO - Iteration 11 of 20
2025-12-19 09:43:48,468 - harmonypy - INFO - Iteration 12 of 20
2025-12-19 09:43:49,388 - harmonypy - INFO - Iteration 13 of 20
2025-12-19 09:43:50,300 - harmonypy - INFO - Iteration 14 of 20
2025-12-19 09:43:51,118 - harmonypy - INFO - Iteration 15 of 20
2025-12-19 09:43:52,079 - harmonypy - INFO - Converged after 15 iterations
rna_meta = adata_rna.obs
rna_meta["gex_cb"] = rna_meta.index
rna_meta["gex_cb"] =[re.sub('-.*','', b) for b in rna_meta["gex_cb"] ]
rna_meta = rna_meta.merge(gex_mc_bc_map)
rna_meta["m_cb"] = rna_meta["m_cb"].astype(str) + "-" + rna_meta["batch"].astype(str)
rna_meta.index = rna_meta["m_cb"]
rna_meta = rna_meta.loc[adata_met.obs.index,]
adata_met.obs["celltype"] = rna_meta["celltype"]
adata_met.obs["celltype"] = adata_met.obs["celltype"].astype('category')plt.rcParams['figure.dpi'] = 150
plt.rcParams['figure.figsize'] = (3,3)
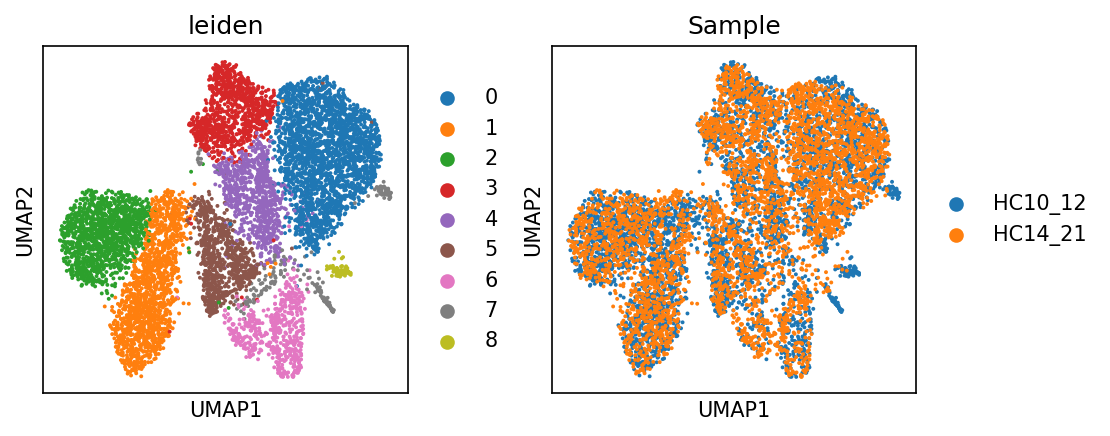
sc.pl.umap(adata_met, color = ['leiden', 'Sample'])cax = scatter(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:394: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
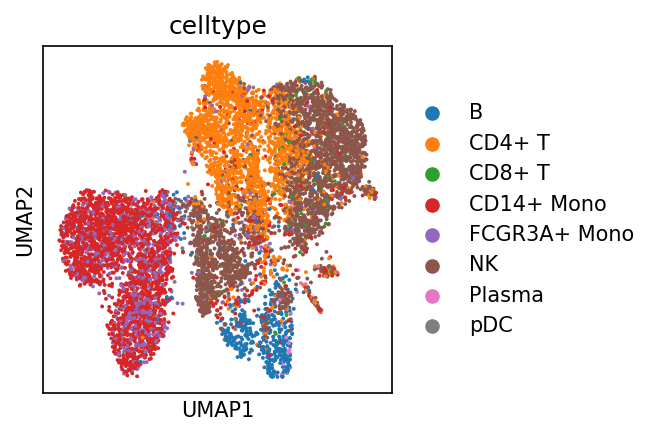
sc.pl.umap(adata_met, color = ['celltype'])cax = scatter(
Cell Composition Proportions
Shows relative abundance of cell types in different samples. Stacked bar charts intuitively reflect inter-sample heterogeneity in cell composition.
Sample-Cell Type Proportion Distribution Plot
This plot shows the proportion of different methylation cell types in each sample.
- X-axis: Represents different samples (SAMple).
- Y-axis: Represents cell proportion (Proportion, 0-1.0).
- Color: Different colored blocks represent different methylation cell types.
def plot_bar_fraction_data(adata, x_key, y_key):
df = adata_met.obs[[x_key, y_key]].copy()
df[y_key] = df[y_key].astype('category')
df[x_key] = df[x_key].astype('category')
counts = df.groupby([x_key, y_key]).size().reset_index(name='count')
totals = counts.groupby(x_key)['count'].transform('sum')
counts['prop'] = counts['count'] / totals
prop_pivot = counts.pivot(index=x_key, columns=y_key, values='prop').fillna(0)
clusters = list(prop_pivot.columns)
if f'{y_key}_colors' in adata_met.uns.keys():
palette = adata.uns['celltype_colors']
palette = sc.pl.palettes.default_20
color_map = {c: palette[i % len(palette)] for i, c in enumerate(clusters)}
bar_colors = [color_map[c] for c in clusters]
return prop_pivot, bar_colors
# Calculate proportion of each cluster per sample and plot stacked bar chart
y_key = 'celltype'
prop_pivot, bar_colors = plot_bar_fraction_data(adata_met, x_key = "Sample", y_key = y_key)
plt.rcParams['figure.dpi'] = 150
plt.rcParams['figure.figsize'] = (3,3)
fig, ax = plt.subplots()
prop_pivot.plot(kind='bar', stacked=True, color=bar_colors, ax=ax)
ax.set_ylabel('Proportion')
ax.set_xlabel("Sample")
ax.legend(title=y_key, bbox_to_anchor=(1, 1), loc='upper left', frameon=False, fontsize=7, markerscale=0.5, handlelength=1.0, handletextpad=0.4, borderaxespad=0.4)
plt.tight_layout()
#prop_pivot.to_csv(f'{sample_col}_{y_key}_frac.csv')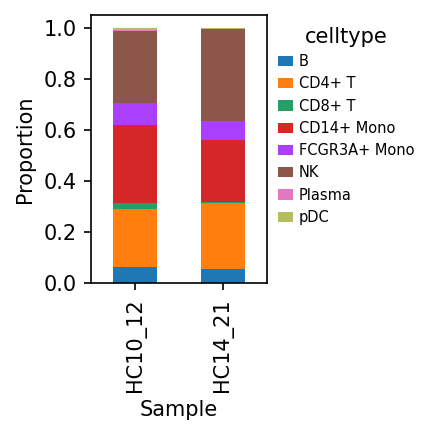
Spatial Distribution Visualization of QC Metrics
QC Metrics Distribution Analysis: UMAP Visualization
Visualization Purpose
By visualizing key QC metrics in UMAP space, we can intuitively assess data quality distribution patterns across cell populations, identify potential technical bias regions, and verify reliability of cell subpopulation partitioning.
total_cpg_number | Total CpG Sites
Interpretation: Color gradient from dark blue to bright yellow reflects the total number of methylation sites captured per cell.
- Bright Yellow Regions: Information-rich, comprehensive methylation landscape
- Dark Blue Regions: Sparse data, interpret biological significance with caution
- Abnormal Patterns: Consistently low values in specific cell clusters may suggest technical issues
reads_counts | Sequencing Depth
Interpretation: Shows the number of unique aligned sequences per cell.
- Depth and Accuracy: Sufficient sequencing depth guarantees methylation quantification accuracy
- QC Standard: Systematically low-depth regions may require technical filtering
genome_cov | Genome Coverage (0-1)
Interpretation: Quantifies the proportion of genomic regions covered by sequencing data.
- Comprehensiveness: High coverage represents a more complete epigenetic profile
- Technical Validation: Abnormally low coverage may point to cell lysis or amplification issues
CpG% | CpG Methylation Rate (0-100%)
Interpretation: Shows overall methylation level of CpG sites.
- Functional Association: Usually associated with gene silencing and chromatin state
- Cell State: Cells in different differentiation or functional states may show characteristic patterns
- Quality Relation: Extreme high/low values may reflect technical anomalies rather than biological differences
CH% | CH Non-CpG Methylation Rate (0-100%)
Interpretation: Reveals modification levels of non-canonical methylation sites.
- Biological Specialization: Has regulatory significance in specific types like neurons, stem cells
- Signal Identification: Complementary to CpG% patterns, providing a more comprehensive epigenetic view
- Baseline Reference: Usually maintained at low levels in mammals; abnormally high values need careful validation
def compute_cg_ch_level(df):
# comput cpg methylation levels and ch methylation levels
cg_cov_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_cov')]
cg_mc_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_mc')]
ch_cov_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_cov')]
ch_mc_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_mc')]
df['CpG_cov'] = df[cg_cov_col].sum(axis=1)
df['CpG_mc'] = df[cg_mc_col].sum(axis=1)
df['CpG%'] = round(df['CpG_mc'] * 100 / df['CpG_cov'], 2)
df['CH_cov'] = df[ch_cov_col].sum(axis=1)
df['CH_mc'] = df[ch_mc_col].sum(axis=1)
df['CH%'] = round(df['CH_mc'] * 100 / df['CH_cov'], 2)
return df
suffix_map = {f'{value}': index for index, value in enumerate(samples)}
meta_list = list()
keep_col = ['barcode', 'total_cpg_number', 'genome_cov', 'reads_counts', 'CpG%', 'CH%']
for i in samples:
s_cells_meta = pd.read_csv(os.path.join(f'{i}', f'{i}_methy', 'step3', 'split_bams','merged', f'{i}_cells.csv'), header = 0)
s_cells_meta['barcode'] = [b.replace('_allc.gz','') for b in s_cells_meta['cell_barcode']]
s_reads_counts = pd.read_csv(os.path.join(f'{i}', f'{i}_methy', 'step3', 'split_bams','merged', 'filtered_barcode_reads_counts.csv'), header = 0)
s_merged_df = s_cells_meta.merge(s_reads_counts, how = 'inner', left_on = 'barcode', right_on = 'barcode')
s_merged_df = compute_cg_ch_level(s_merged_df)
s_merged_df = s_merged_df[keep_col]
if len(samples) > 1:
s_merged_df['barcode'] = [f'{b}-{suffix_map[i]}'for b in s_merged_df['barcode']]
meta_list.append(s_merged_df)
adata_met.obs = adata_met.obs.merge(
pd.concat(meta_list).set_index('barcode'),
how = 'left',
left_index=True,
right_index = True)ch_cov_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_cov')]
/tmp/ipykernel_143/2478518388.py:6: UserWarning: This pattern is interpreted as a regular expression, and has match groups. To actually get the groups, use str.extract.
ch_mc_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_mc')]
/tmp/ipykernel_143/2478518388.py:5: UserWarning: This pattern is interpreted as a regular expression, and has match groups. To actually get the groups, use str.extract.
ch_cov_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_cov')]
/tmp/ipykernel_143/2478518388.py:6: UserWarning: This pattern is interpreted as a regular expression, and has match groups. To actually get the groups, use str.extract.
ch_mc_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_mc')]
plt.rcParams['figure.dpi'] = 150
plt.rcParams['figure.figsize'] = (3,3)
sc.pl.umap(adata_met, color = ['total_cpg_number','reads_counts','genome_cov'], cmap = 'inferno')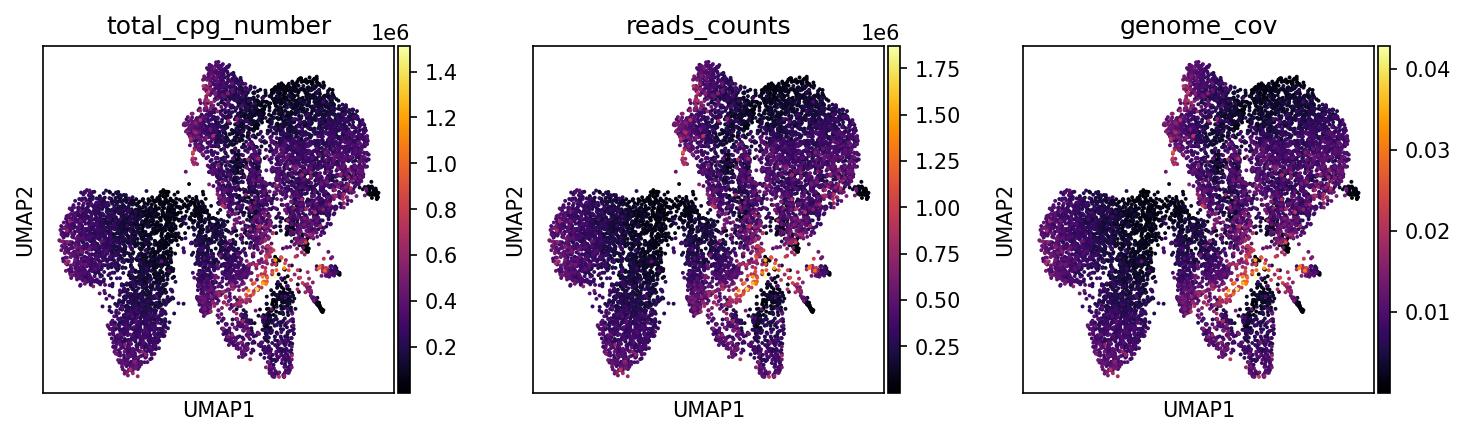
plt.rcParams['figure.dpi'] = 150
plt.rcParams['figure.figsize'] = (3,3)
sc.pl.umap(adata_met, color = ['CpG%','CH%'], cmap = 'inferno')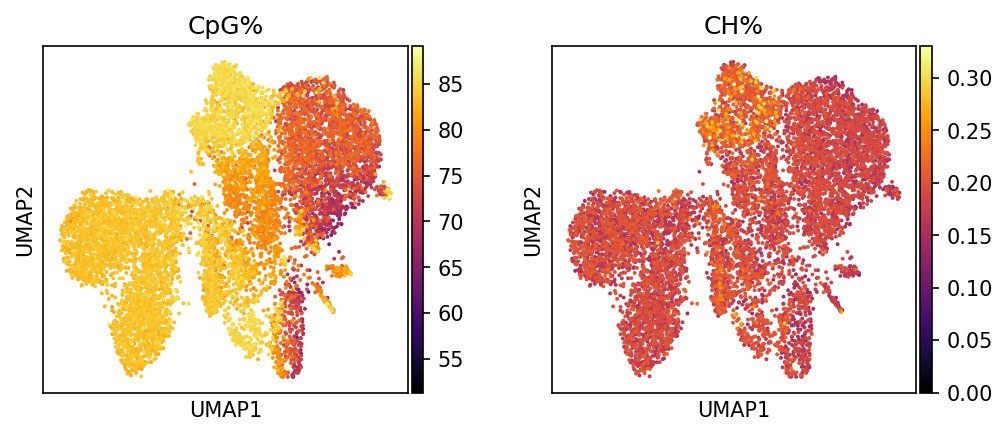
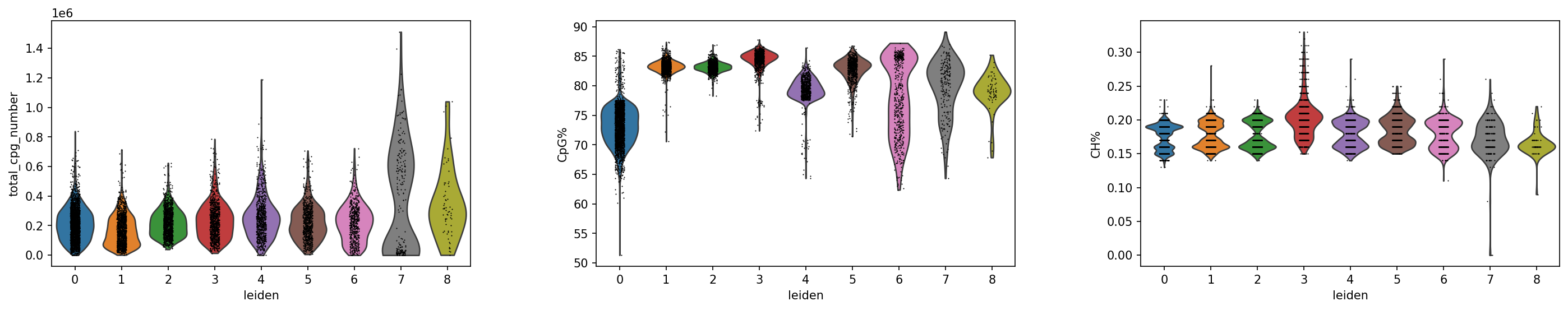
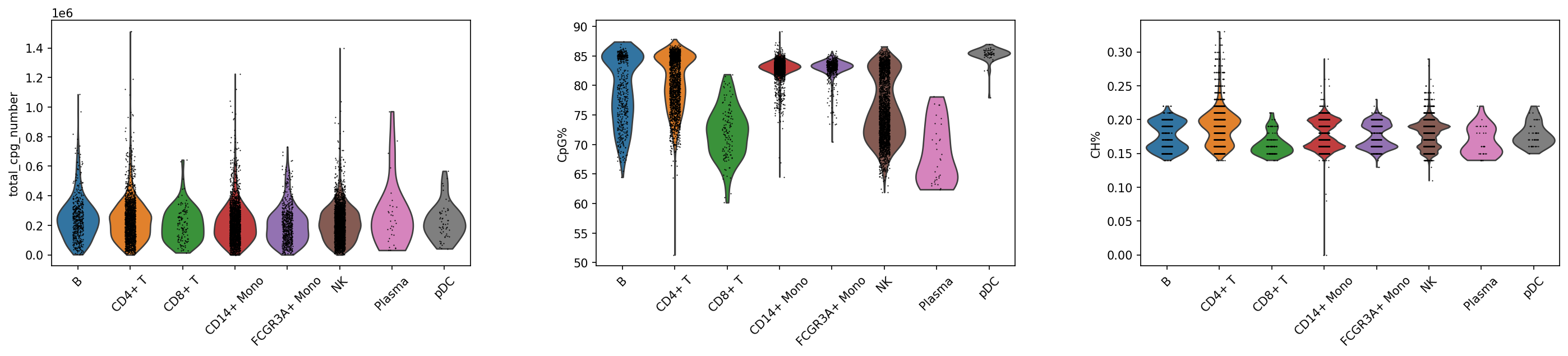
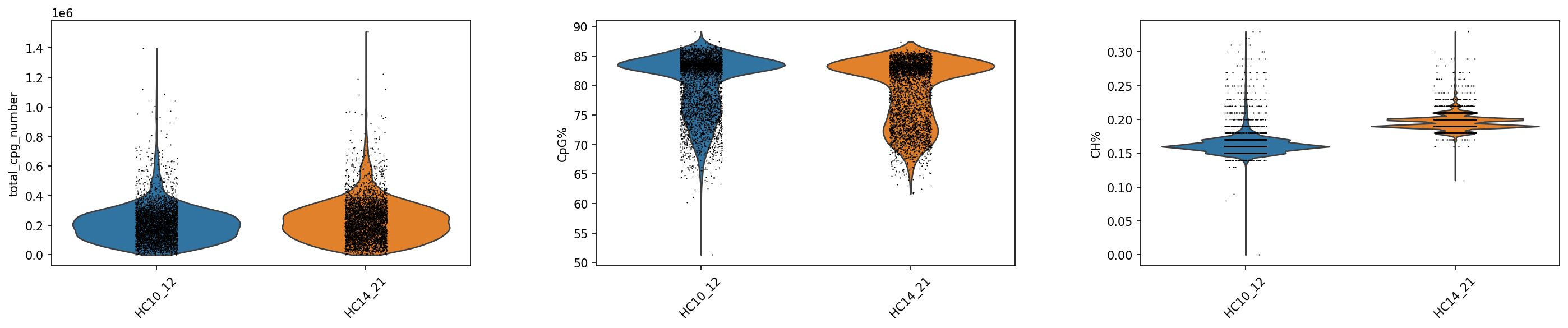
QC Metrics Distribution Analysis: Violin Plot Visualization
Visualization Purpose
Using Violin Plots to intuitively display distribution characteristics of key QC metrics across different cell groups, assessing data quality uniformity, identifying potential batch effects or technical biases, and ensuring reliability of subsequent biological findings.
CpG Site Count Distribution
- Ideal Pattern: Similar distribution across clusters, medians close
- Abnormal Warning: Significantly lower site counts in specific clusters
- Biological Significance: Reflects cell capture efficiency and DNA quality
CpG Methylation Rate Distribution
- Expected Range: Usually maintains a relatively stable baseline level in mammalian cells
- Inter-cluster Differences: Different cell types may show characteristic methylation levels
- QC Standard: Abnormally low/high values may indicate technical issues
CH Methylation Rate Distribution
- Baseline Characteristics: Usually far lower than CpG methylation rate
- Biological Specificity: May be elevated in specific types like neurons, embryonic stem cells
- Technical Differentiation: Need to distinguish real biological features from technical biases
plt.rcParams['figure.figsize'] = (6,4)
sc.pl.violin(adata_met, keys=['total_cpg_number', 'CpG%', 'CH%'], groupby='leiden')
#_ = sc.pl.violin(adata_met, keys=['total_cpg_number', 'CpG%', 'CH%'], groupby='leiden', save = '_leiden_cpg_ch.png', show = False)Passing \`palette\` without assigning \`hue\` is deprecated and will be removed in v0.14.0. Assign the \`x\` variable to \`hue\` and set \`legend=False\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The \`scale\` parameter has been renamed and will be removed in v0.15.0. Pass \`density_norm='width'\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
Passing \`palette\` without assigning \`hue\` is deprecated and will be removed in v0.14.0. Assign the \`x\` variable to \`hue\` and set \`legend=False\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The \`scale\` parameter has been renamed and will be removed in v0.15.0. Pass \`density_norm='width'\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
Passing \`palette\` without assigning \`hue\` is deprecated and will be removed in v0.14.0. Assign the \`x\` variable to \`hue\` and set \`legend=False\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The \`scale\` parameter has been renamed and will be removed in v0.15.0. Pass \`density_norm='width'\` for the same effect.
ax = sns.violinplot(
plt.rcParams['figure.figsize'] = (6,4)
sc.pl.violin(adata_met, keys=['total_cpg_number', 'CpG%', 'CH%'], groupby="celltype",rotation = 45)
#_ = sc.pl.violin(adata_met, keys=['total_cpg_number', 'CpG%', 'CH%'], groupby=anno_col, save = f'_{anno_col}_cpg_ch.png', rotation = 45,show = False)Passing \`palette\` without assigning \`hue\` is deprecated and will be removed in v0.14.0. Assign the \`x\` variable to \`hue\` and set \`legend=False\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The \`scale\` parameter has been renamed and will be removed in v0.15.0. Pass \`density_norm='width'\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
Passing \`palette\` without assigning \`hue\` is deprecated and will be removed in v0.14.0. Assign the \`x\` variable to \`hue\` and set \`legend=False\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The \`scale\` parameter has been renamed and will be removed in v0.15.0. Pass \`density_norm='width'\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
Passing \`palette\` without assigning \`hue\` is deprecated and will be removed in v0.14.0. Assign the \`x\` variable to \`hue\` and set \`legend=False\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The \`scale\` parameter has been renamed and will be removed in v0.15.0. Pass \`density_norm='width'\` for the same effect.
ax = sns.violinplot(
plt.rcParams['figure.figsize'] = (6,4)
sc.pl.violin(adata_met, keys=['total_cpg_number', 'CpG%', 'CH%'], groupby="Sample",rotation = 45)
#_ = sc.pl.violin(adata_met, keys=['total_cpg_number', 'CpG%', 'CH%'], groupby=sample_col, save = f'_{sample_col}_cpg_ch.png',rotation = 45, show = False)Passing \`palette\` without assigning \`hue\` is deprecated and will be removed in v0.14.0. Assign the \`x\` variable to \`hue\` and set \`legend=False\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The \`scale\` parameter has been renamed and will be removed in v0.15.0. Pass \`density_norm='width'\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
Passing \`palette\` without assigning \`hue\` is deprecated and will be removed in v0.14.0. Assign the \`x\` variable to \`hue\` and set \`legend=False\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The \`scale\` parameter has been renamed and will be removed in v0.15.0. Pass \`density_norm='width'\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
Passing \`palette\` without assigning \`hue\` is deprecated and will be removed in v0.14.0. Assign the \`x\` variable to \`hue\` and set \`legend=False\` for the same effect.
ax = sns.violinplot(
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_anndata.py:839: FutureWarning:
The \`scale\` parameter has been renamed and will be removed in v0.15.0. Pass \`density_norm='width'\` for the same effect.
ax = sns.violinplot(
adata_met.write_h5ad('adata_met.h5ad')