Single-cell Gene Expression Dynamics: Pseudotime Gene Expression Curve Fitting
Time: 3 min
Words: 526 words
Updated: 2026-02-27
Reads: 0 times
Environment Setup
R
# Load required R packages
library(monocle)
library(Seurat)
library(dplyr)
library(tidyverse)
library(ggplot2)
library(ggsci)Data Loading
R
# Read data
gbm_cds <- readRDS("data/AY1740561864613/advanced_analysis/58047/output/monocle2/monocle_final.rds") ### Monocle2 analysis results
rep <- as.data.frame(pData(gbm_cds)) # Read phenotype information
gene_all <- row.names(gbm_cds@assayData$exprs) # Get all genes in gbm_cdsData Preprocessing
R
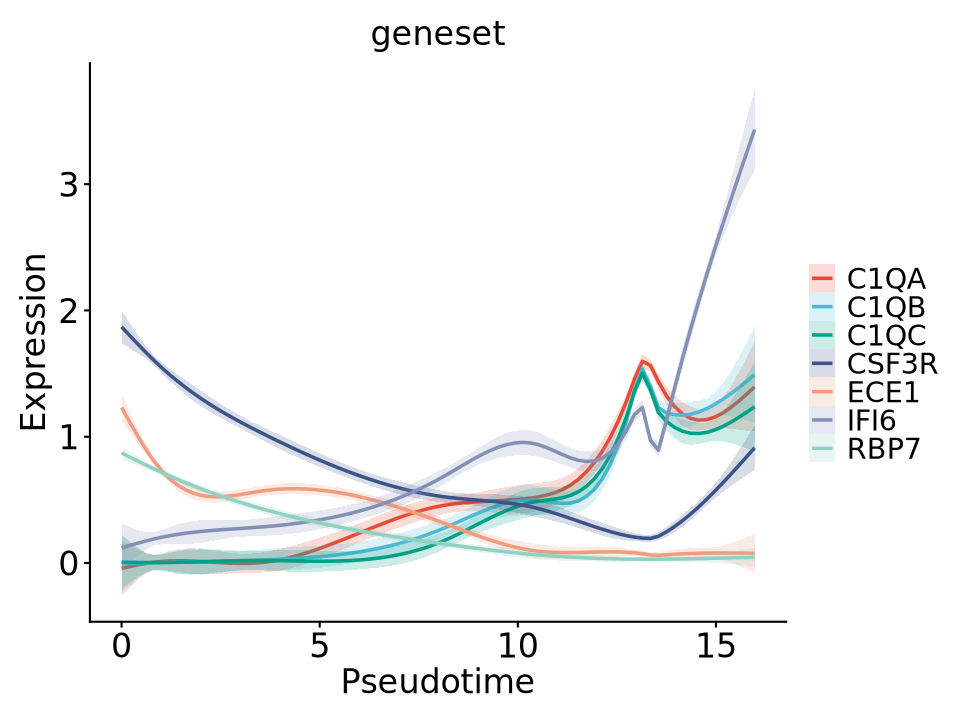
gene_ad <- c("RBP7","ECE1","C1QA","C1QC","C1QB","IFI6","CSF3R") # Define genes to display (max 10)R
# Intersect gene_ad with genes_all
gene_se <- vector()
for(i in gene_ad){
if(i %in% gene_all){
gene_se <- c(gene_se,i)
}
}R
# Extract gene expression levels
for(i in gene_se){
gene_in <- names(pData(gbm_cds))
if (i %in% gene_in){
next
}else{
tmp <- gbm_cds@assayData$exprs[i,] %>% as.data.frame
names(tmp) <- i
tmp <- tmp %>% mutate(barcode=rownames(tmp))
pData(gbm_cds) <- pData(gbm_cds) %>% inner_join(tmp,by="barcode")
rownames(pData(gbm_cds)) <- pData(gbm_cds)$barcode
}
}R
# Merge expression data with pseudotime info
data_plot <- pData(gbm_cds) %>% select(Pseudotime,all_of(gene_se)) %>%
pivot_longer(cols = -Pseudotime,names_to = "Gene",values_to = "Expression")Pseudotime Curve Plotting
R
# Set plot output dimensions (for interactive environments like Jupyter)
# repr.plot.height: Plot height 6 inches
# repr.plot.width: Plot width 8 inches
options(repr.plot.height=6, repr.plot.width=8)
# Create ggplot object
ggplot(data = data_plot, mapping = aes(x = Pseudotime,
y = Expression,
color = Gene, # Color by gene
fill = Gene)) + # Fill color by gene
# Use Nature Publishing Group color scheme (npg)
ggsci::scale_color_npg(name = "") + # Discrete color scale
ggsci::scale_fill_npg(name = "") + # Discrete fill color scale
# Add GAM smoothing curve (confidence interval band)
# method: Use Generalized Additive Model
# formula: Cubic spline basis function
# alpha: Confidence interval transparency (0-1)
# size: Curve line width
geom_smooth(method = "gam",
formula = y ~ s(x, bs = "cs"),
alpha = 0.2,
size = 1) +
# Apply cowplot theme (concise scientific style)
cowplot::theme_cowplot() +
# Customize theme details
theme(
axis.text.x = element_text(size = 20), # X-axis tick label size
axis.text.y = element_text(size = 20), # Y-axis tick label size
text = element_text(size = 20, family = "ArialMT"), # Global font settings
plot.margin = unit(c(1, 1, 1, 1), "char"), # Plot margins (character units)
plot.title = element_text(
hjust = 0.5, # Center title
size = 20, # Title font size
family = "ArialMT", # Font family
face = "plain" # No bold
),
axis.line = element_line(
linetype = 1, # Solid line type
color = "black", # Axis line color
linewidth = 0.6 # Axis line width
),
axis.ticks = element_line(
linetype = 1, # Tick line type
color = "black", # Tick line color
linewidth = 0.6 # Tick line width
)
# legend.position = "none" # Hide legend (currently commented out)
) +
# Axis Label Settings
ylab("Expression") + # Y-axis label (Gene Expression)
xlab("Pseudotime") + # X-axis label (Pseudotime Analysis Result)
ggtitle("geneset") # Main Title
# Save Output (Publication Quality)
# filename: Output filename
# width: PDF width 9 inches (approx 22.86cm)
# height: PDF height 8 inches (approx 20.32cm)
ggsave("expression.pdf", width = 9, height = 8)