SeekArc Single-Cell Multi-Omics (RNA+ATAC) Single Sample Analysis Tutorial (WNN)
Environment Preparation
Load R Packages
Please select the common_r environment for this integration tutorial
# Load necessary R packages
suppressPackageStartupMessages({
library(Seurat)
library(Signac)
library(EnsDb.Hsapiens.v86, lib.loc = "/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/python3.10/lib/R/library")
library(BSgenome.Hsapiens.UCSC.hg38,lib = "/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/python3.10/lib/R/library")
library(biovizBase, lib = "/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/python3.10/lib/R/library")
#library(BSgenome.Mmusculus.UCSC.mm10)
#library(EnsDb.Mmusculus.v79)
library(dplyr)
library(ggplot2)
library(patchwork)
library(harmony)
})
# Set random seed
set.seed(1234)
# Set Seurat options (Note: 8000 * 1024^2 is actually 8GB)
options(future.globals.maxSize = 8000 * 1024^2) # 8GB# Define color scheme
my36colors <-c( '#E5D2DD', '#53A85F', '#F1BB72', '#F3B1A0', '#D6E7A3', '#57C3F3', '#476D87',
'#E95C59', '#E59CC4', '#AB3282', '#23452F', '#BD956A', '#8C549C', '#585658',
'#9FA3A8', '#E0D4CA', '#5F3D69', '#C5DEBA', '#58A4C3', '#E4C755', '#F7F398',
'#AA9A59', '#E63863', '#E39A35', '#C1E6F3', '#6778AE', '#91D0BE', '#B53E2B',
'#712820', '#DCC1DD', '#CCE0F5', '#CCC9E6', '#625D9E', '#68A180', '#3A6963',
'#968175', "#6495ED", "#FFC1C1",'#f1ac9d','#f06966','#dee2d1','#6abe83','#39BAE8','#B9EDF8','#221a12',
'#b8d00a','#74828F','#96C0CE','#E95D22','#017890')Get Gene Annotation Information
We will obtain genome annotation information (gene positions, transcripts, exons, TSS, etc.) for the corresponding species from the EnsDb database. The specific species needs to be changed according to the data. This information is used for:
- Calculating ATAC TSS enrichment (determining if open chromatin is more concentrated near transcription start sites)
- Constructing gene activity matrix (mapping peak signals to genes)
- Peak annotation and functional analysis
Notes:
- Species must match reference genome version (e.g.,
EnsDb.Hsapiens.v86for hg38,EnsDb.Mmusculus.v75for mm10) - Consistent chromosome naming style (e.g., using prefixes like
chr1,chr2)
# Get gene annotation information (silence warnings and messages)
suppressWarnings({
suppressMessages({
annotation <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
seqlevels(annotation) <- paste0('chr', seqlevels(annotation))
genome(annotation) <- 'hg38'
})
})Data Reading
This tutorial provides two different data input methods to meet different user data acquisition needs. Choose the one that suits you:
Cloud Platform RDS File Reading
Data Characteristics:
- RDS file is a standard Seurat object file
- Can be directly used for subsequent downstream analysis, or expression matrix can be extracted for re-integration
Applicable Scenarios:
- When you cannot obtain the standard
filtered_feature_bc_matrixexpression matrix - When you want to use existing data on the cloud platform for learning
- When you need to quickly re-perform basic analysis
Notes:
- For specific data mounting and RDS file reading, please refer to the Jupyter usage tutorial
For example, the following project data /home/demo-SeekGene-com/workspace/data/AY1752565399550/
# For example, the following project data /home/demo-seekgene-com/workspace/data/AY1752565399550/
#input <- readRDS("/home/demo-seekgene-com/workspace/data/AY1752565399550/input.rds")
#meta <- read.table("/home/demo-seekgene-com/workspace/data/AY1752565399550/meta.tsv", header=TRUE, sep="\t", row.names = 1)
# Extract genome annotation file from original RDS data
#annotations <- Annotation(input)
#seu <- CreateSeuratObject(counts = input@assay$RNA@counts, meta.data=meta)
#seu[["ATAC"]] <- CreateChromatinAssay(seu, counts = input@assay$ATAC@counts, fragments = input@assay$ATAC@fragments)
#Annotation(seu) <- annotations
#rm(input)
#gc()Standard filtered_XXXX_bc_matrix File Reading
Applicable Scenarios:
- When you have standard gene expression matrix and peaks open matrix files
- When you want to independently complete basic analysis of single-cell multi-omics (SeekArc) data
- When you need a complete workflow from raw data to analysis results
Note:
- Please ensure the file structure is as follows:
The data directory structure must meet the following requirements:
- The sample folder name is the sample ID, such as S127.
- The sample folder contains the following files:
filtered_feature_bc_matrix: scRNA-seq expression matrix folder, containingbarcodes.tsv.gz,features.tsv.gz, andmatrix.mtx.gzfiles.filtered_peaks_bc_matrix: ATAC peak open matrix folder, containingbarcodes.tsv.gz,features.tsv.gz, andmatrix.mtx.gzfiles.{Sample ID}_A_fragments.tsv.gz: ATAC fragments file, such as S127_A_fragments.tsv.gz.{Sample ID}_A_fragments.tsv.gz.tbi: ATAC fragments index file, such as S127_A_fragments.tsv.gz.tbi.
The specific folder structure is as follows:
├── S127/
│ ├── filtered_feature_bc_matrix/ (scRNA-seq expression matrix)
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── filtered_peaks_bc_matrix/ (ATAC peak open matrix)
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── S127_A_fragments.tsv.gz (ATAC fragments file)
│ └── S127_A_fragments.tsv.gz.tbi
# load the RNA and ATAC data
RNA_counts <- Read10X("./S127/filtered_feature_bc_matrix/")
ATAC_counts <- Read10X("./S127/filtered_peaks_bc_matrix/")
fragpath <- "./S127/S127_A_fragments.tsv.gz"
# create a Seurat object containing the RNA adata
seu <- CreateSeuratObject(
counts = RNA_counts,
assay = "RNA"
)
# create ATAC assay and add it to the object
seu[["ATAC"]] <- CreateChromatinAssay(
counts = ATAC_counts,
sep = c(":", "-"),
fragments = fragpath,
annotation = annotation
)Data Quality Control
Calculation of Quality Control Metrics
RNA QC Metrics:
percent.mt: Percentage of mitochondrial genes (usually <20%)nfeature_RNA: Number of RNA features (usually between 200-10000)
ATAC QC Metrics:
TSS.enrichment: TSS enrichment score (usually >2)nucleosome_signal: Nucleosome signal (lower is better)nCount_ATAC: Total ATAC count (usually between 1000-10000)
# Perform quality control for each sample
suppressWarnings({
suppressMessages({
# RNA QC metrics
seu[["percent.mt"]] <- PercentageFeatureSet(seu, pattern = "^MT-")
# ATAC QC metrics
DefaultAssay(seu) <- "ATAC"
# Calculate TSS enrichment score
seu <- TSSEnrichment(object = seu, fast = FALSE)
# Calculate nucleosome signal
seu <- NucleosomeSignal(object = seu)
})
})Visualization of Quality Control Metrics
Use violin plots to view the distribution/outliers of QC metrics and determine appropriate thresholds:
Suggestions:
- Observe if there are obvious long tails or bimodal distributions
- Try multiple sets of thresholds and compare if downstream clustering/UMAP is clearer
# Visualize QC metrics. This cell is optional.
options(repr.plot.width = 17, repr.plot.height = 10)
suppressWarnings({
p1=DensityScatter(seu, x = 'nCount_ATAC', y = 'TSS.enrichment', log_x = TRUE, quantiles = TRUE)
p2=VlnPlot(
object = seu,
features = c('nCount_ATAC', 'TSS.enrichment', 'nucleosome_signal',"nFeature_RNA", "nCount_RNA", "percent.mt"),
pt.size = 0.1,
ncol = 6
)
print(p1 / p2)
})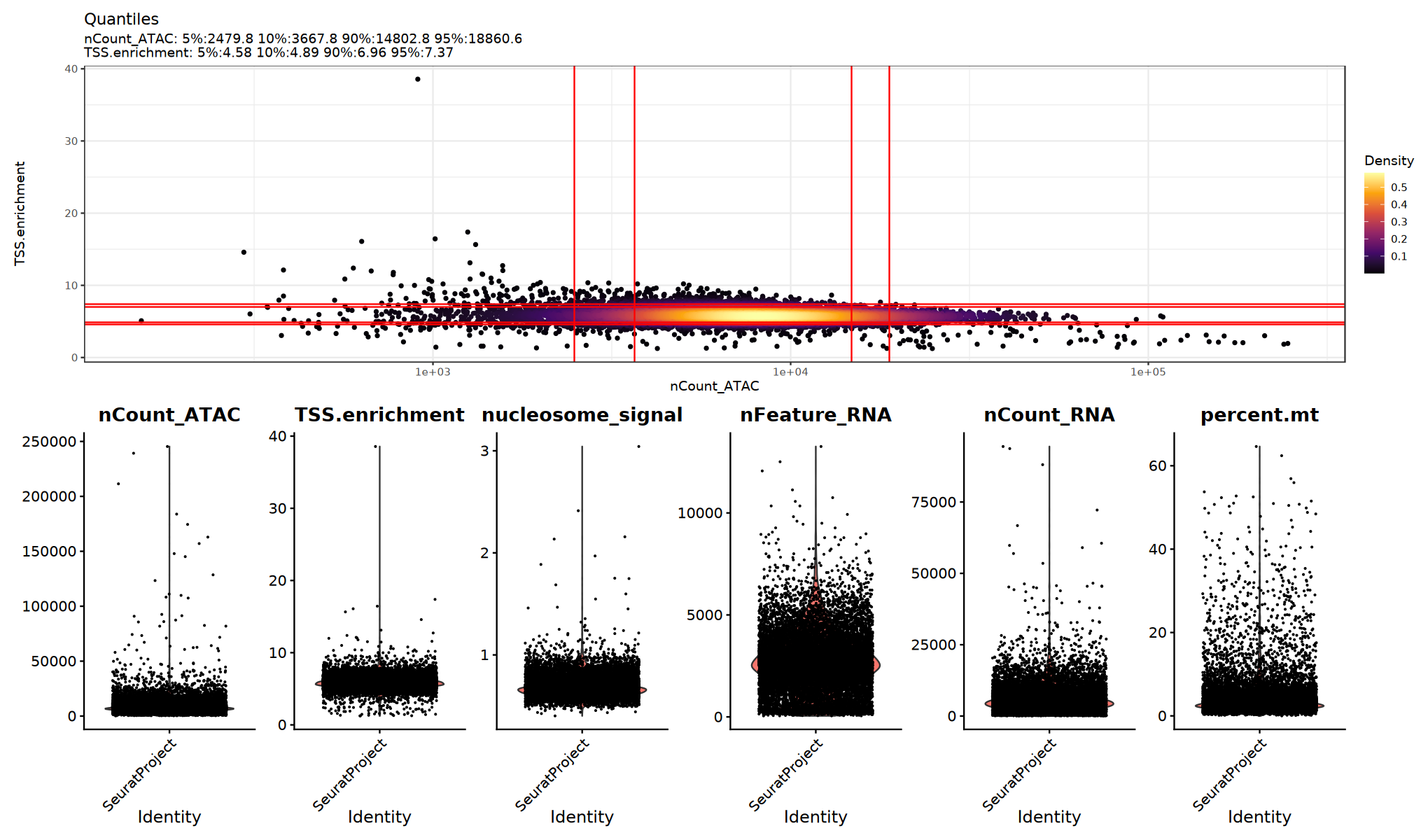
Filtering Low Quality Cells
Filter low quality cells based on QC metrics. Specific thresholds should be adjusted according to data characteristics and violin plot distributions.
# Quality Control Filtering
cells_before <- ncol(seu)
seu <- subset(seu,
subset = nFeature_RNA > 200 &
nFeature_RNA < 8000 &
nCount_RNA > 500 &
nCount_RNA < 30000 &
percent.mt < 20 &
nCount_ATAC > 500 &
nCount_ATAC < 100000 &
TSS.enrichment > 1 &
nucleosome_signal < 2
)
cells_after <- ncol(seu)Data Normalization
RNA Data Normalization and Linear Dimensionality Reduction
Normalization Method: By default, Seurat uses "LogNormalize" global scaling normalization method:
- First normalizes feature expression for each cell by total expression
- Multiplies by a scale factor (default is 10,000)
- Then log-transforms the result
Feature Selection: Identifies a subset of features that exhibit high cell-to-cell variation in the dataset (i.e., they are highly expressed in some cells, and lowly expressed in others)
Data Scaling: Applies a linear transformation ("scaling"), a standard pre-processing step prior to dimensional reduction techniques like PCA
Dimensionality Reduction: Perform PCA on the normalized data
suppressWarnings({
suppressMessages({
DefaultAssay(seu) <- "RNA"
seu <- NormalizeData(seu, assay = "RNA")
seu <- FindVariableFeatures(seu, assay = "RNA", selection.method = "vst", nfeatures = 2000)
seu <- ScaleData(seu, assay = "RNA")
seu <- RunPCA(seu, assay = "RNA", npcs = 50)
})
})ATAC Data Normalization and Linear Dimensionality Reduction
Normalization Method: Signac uses Term Frequency-Inverse Document Frequency (TF-IDF) normalization:
- Normalizes across cells to correct for sequencing depth differences
- Normalizes across peaks to give higher weight to rare peaks
Feature Selection Strategy: Due to the low dynamic range of scATAC-seq data, we cannot perform variable feature selection like in scRNA-seq. Alternatively, we can use FindTopFeatures() to select top n% features (peaks) or remove features present in fewer than n cells.
Dimensionality Reduction: Then perform Singular Value Decomposition (SVD) on the TF-IDF matrix using the selected features.
suppressWarnings({
suppressMessages({
seu <- RunTFIDF(seu, assay = "ATAC")
seu <- FindTopFeatures(seu, assay = "ATAC", min.cutoff = 'q0')
seu <- RunSVD(seu, assay = "ATAC")
})
})Non-linear Dimensionality Reduction and Clustering
Three Clustering Strategies
- RNA Clustering: Based on gene expression similarity
- ATAC Clustering: Based on chromatin accessibility similarity
- WNN Clustering: Integrating both modalities (Recommended)
Result Interpretation
- Different methods may produce different clustering results
- WNN clustering usually reveals more detailed cell subpopulations
- It is recommended to prioritize WNN results for downstream analysis
RNA Dimensionality Reduction and Clustering
RNA Clustering Strategy:
- Independent clustering analysis based on RNA data
- Classifying cells using gene expression patterns
suppressWarnings({
suppressMessages({
seu <- RunUMAP(seu, reduction = "pca", dims = 1:30, assay = "RNA",reduction.name="rnaumap")
seu <- FindNeighbors(seu, reduction = "pca", dims = 1:30, assay = "RNA",graph.name = "rnaneigobr")
seu <- FindClusters(seu, resolution = 0.5, algorithm = 1,graph.name = "rnaneigobr")
})
})Number of nodes: 12694
Number of edges: 119839
Running Louvain algorithm...n Maximum modularity in 10 random starts: 0.9108
Number of communities: 25
Elapsed time: 0 seconds
ATAC Dimensionality Reduction and Clustering
ATAC Clustering Strategy:
- Independent clustering analysis based on ATAC data
- Classifying cells using chromatin accessibility patterns
suppressWarnings({
suppressMessages({
seu <- RunUMAP(seu, reduction = "lsi", dims = 1:30, assay = "ATAC",reduction.name="atacumap")
seu <- FindNeighbors(seu, reduction = "lsi", dims = 1:30, assay = "ATAC",graph.name = "atacneigobr")
seu <- FindClusters(seu, resolution = 0.5, algorithm = 1,graph.name = "atacneigobr")
})
})Number of nodes: 12694
Number of edges: 120530
Running Louvain algorithm...n Maximum modularity in 10 random starts: 0.8927
Number of communities: 19
Elapsed time: 0 seconds
Weighted Nearest Neighbor (WNN) Analysis
WNN Integrated Clustering Strategy:
- Weighted nearest neighbor analysis combining RNA and ATAC data
- Provides more accurate cell type identification
suppressWarnings({
suppressMessages({
seu <- FindMultiModalNeighbors(seu, reduction.list = list("pca", "lsi"), dims.list = list(1:30, 1:30))
# Clustering based on WNN
seu <- FindClusters(seu, graph.name = "wknn", resolution = 0.5)
# WNN UMAP
seu <- RunUMAP(seu, nn.name = "weighted.nn", reduction.name = "wnn.umap")
})
})Number of nodes: 12694
Number of edges: 191058
Running Louvain algorithm...n Maximum modularity in 10 random starts: 0.8993
Number of communities: 20
Elapsed time: 0 seconds
Visualization
# Compare clustering results of different methods
p1 <- DimPlot(seu, reduction = "rnaumap", group.by = "rnaneigobr_res.0.5",label=T, cols = my36colors) + ggtitle("RNA")
p2 <- DimPlot(seu, reduction = "atacumap", group.by = "atacneigobr_res.0.5",label=T, cols = my36colors) + ggtitle("ATAC")
p3 <- DimPlot(seu, reduction = "wnn.umap", group.by = "wknn_res.0.5",label=T, cols = my36colors) + ggtitle("WNN")
options(repr.plot.width=23, repr.plot.height=6)
print(p1 + p2 + p3)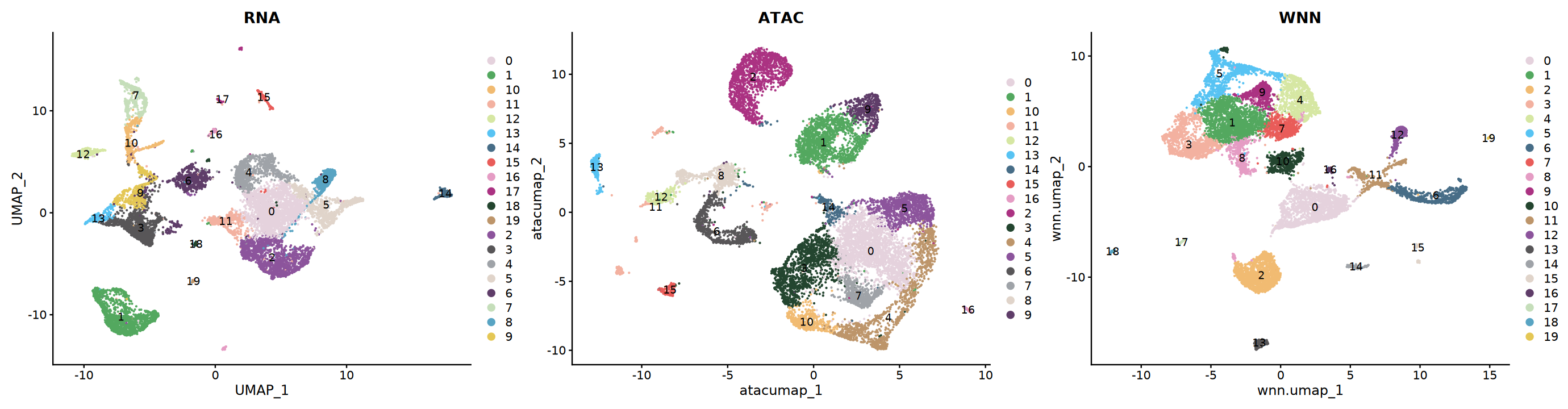
Cell Type Annotation
Marker Genes
Collect marker gene sets for different cell types based on tissue type. This example data is from the eye. Below are cell types in the eye and their corresponding marker genes. Use dot plots to visualize which cell type markers are highly expressed in different clusters.
eye_marker_integrated <- list(
# ========== Photoreceptor Cells ==========
"Rod_Photoreceptors" = c("RHO", "PDE6B", "CNGB1", "PDE6A", "NR2E3", "REEP6",
"RCVRN", "SAG", "NEB", "SLC24A3", "TRPM1"),
"Cone_Photoreceptors" = c("ARR3", "GNAT2", "OPN1SW", "TRPM3"),
# ========== Retinal Neurons ==========
"Bipolar_Cells" = c("VSX1", "OTX2", "GRM6", "PRKCA",
"DLG2", "NRXN3", "RBFOX3", "FSTL4"),
"Amacrine_Cells" = c("GAD1", "GAD2", "C1QL2", "TFAP2B", "SLC32A1"),
"Horizontal_Cells" = c("ONECUT1", "LHX1", "CALB1", "NFIA"),
"Retinal_Ganglion_Cells" = c("RBPMS", "THY1", "NEFL", "POU4F2"),
# ========== Glial Cells ==========
"Muller_Glia" = c("SLC1A3", "RLBP1", "SOX9", "CRYAB"),
"Astrocytes" = c("GFAP", "AQP4", "S100B"),
"Microglia" = c("TMEM119", "C1QA", "AIF1", "CX3CR1", "CD74",
"PTPRC", "CSF1R", "CCL3L1", "SPP1", "P2RY12"),
# ========== Vascular and Support Cells ==========
"Endothelial" = c("FLT1", "PODXL", "PLVAP", "KDR", "EGFL7",
"CLDN5", "PECAM1", "CDH5"),
"Pericytes" = c("PDGFRB", "RGS5", "CSPG4", "STAB1"),
# ========== Epithelial Cells ==========
"RPE" = c("RPE65", "BEST1", "PMEL", "TYRP1", "DCT", "SLC38A11"),
#"Corneal_Epithelial" = c("KRT12", "KRT3"),
"Corneal_Endothelial" = c("SLC4A11", "COL8A2", "ATP1A1"),
"Conjunctival_Epithelial" = c("KRT13", "KRT19", "MUC5AC"),
"Retinal_Progenitors" = c("VSX2", "PAX6","SOX2", "HES1", "NOTCH1"), # Neural retina proliferation stage
# ========== Lens Cells ==========
"Lens_Epithelial" = c("CRYAA", "BFSP1", "MIP"),
"Lens_Fiber" = c("CRYGS", "LIM2", "FN1"),
# ========== Other Cells ==========
"Melanocytes" = c("TYR", "MLANA", "MITF"),
#"Erythrocytes" = c("HBB", "HBA1", "HBA2"),
"ECM" = c("COL1A1", "COL3A1", "COL12A1", "COL1A2", "COL6A3")#,
#"Others" = c("TTN", "CLCN5", "DCC", "MIAT")
)# Set plot size
options(repr.plot.width=25, repr.plot.height=7)
# Draw DotPlot
DefaultAssay(seu)="RNA"
DotPlot(seu,
group.by = "wknn_res.0.5",
features = eye_marker_integrated,
cols = c("#f8f8f8","#ff3472"),
#dot.min = 0.05,
dot.scale = 8)+ # Apply custom colors
RotatedAxis() +
scale_x_discrete("") +
scale_y_discrete("") +
theme(
axis.text.x = element_text(size = 12, face = "bold",
angle = 45, hjust = 1, vjust = 1),
axis.text.y = element_text(size = 12, face = "bold"),
plot.title = element_text(size = 14, face = "bold", hjust = 0.5),
legend.title = element_text(size = 10, face = "bold")
) +
ggtitle("Marker Genes Expression") +
labs(color = "Expression\nLevel") # Modify legend title"The \`facets\` argument of \`facet_grid()\` is deprecated as of ggplot2 2.2.0.
ℹ Please use the \`rows\` argument instead.
ℹ The deprecated feature was likely used in the Seurat package.
Please report the issue at
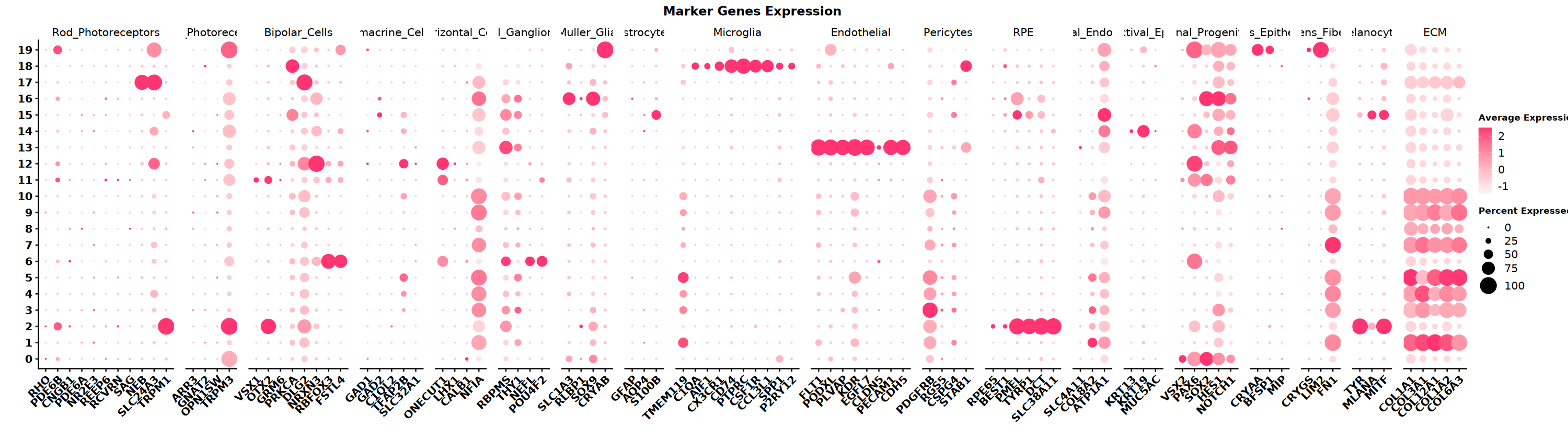
** Notes ** SeekArc single-cell dual-omics cell annotation is generally based on WNN multimodal dimensionality reduction, annotating based on marker gene expression in wknn clustering results
options(repr.plot.width=10, repr.plot.height=8)
DimPlot(seu, reduction = "wnn.umap", group.by = "wknn_res.0.5", cols = my36colors,label = T)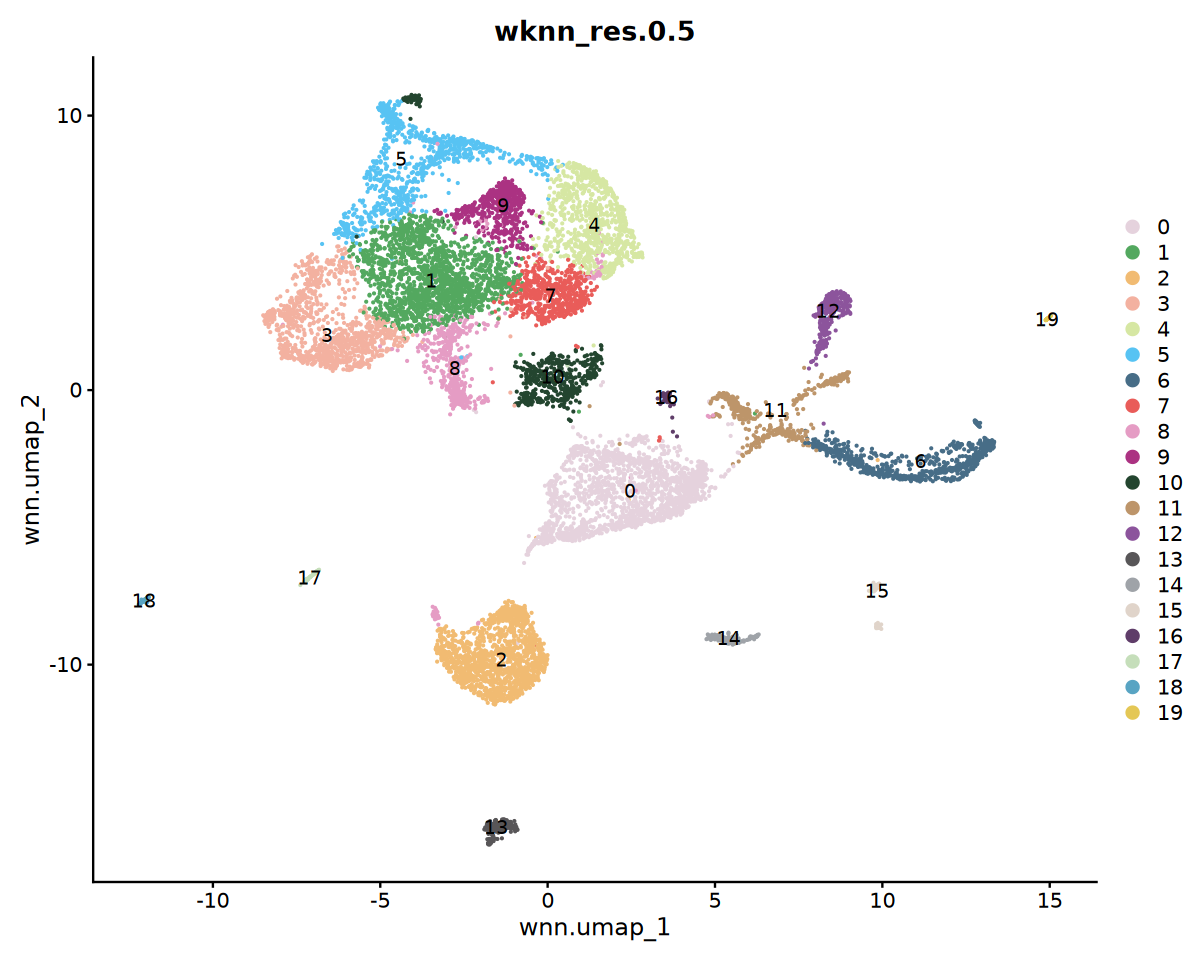
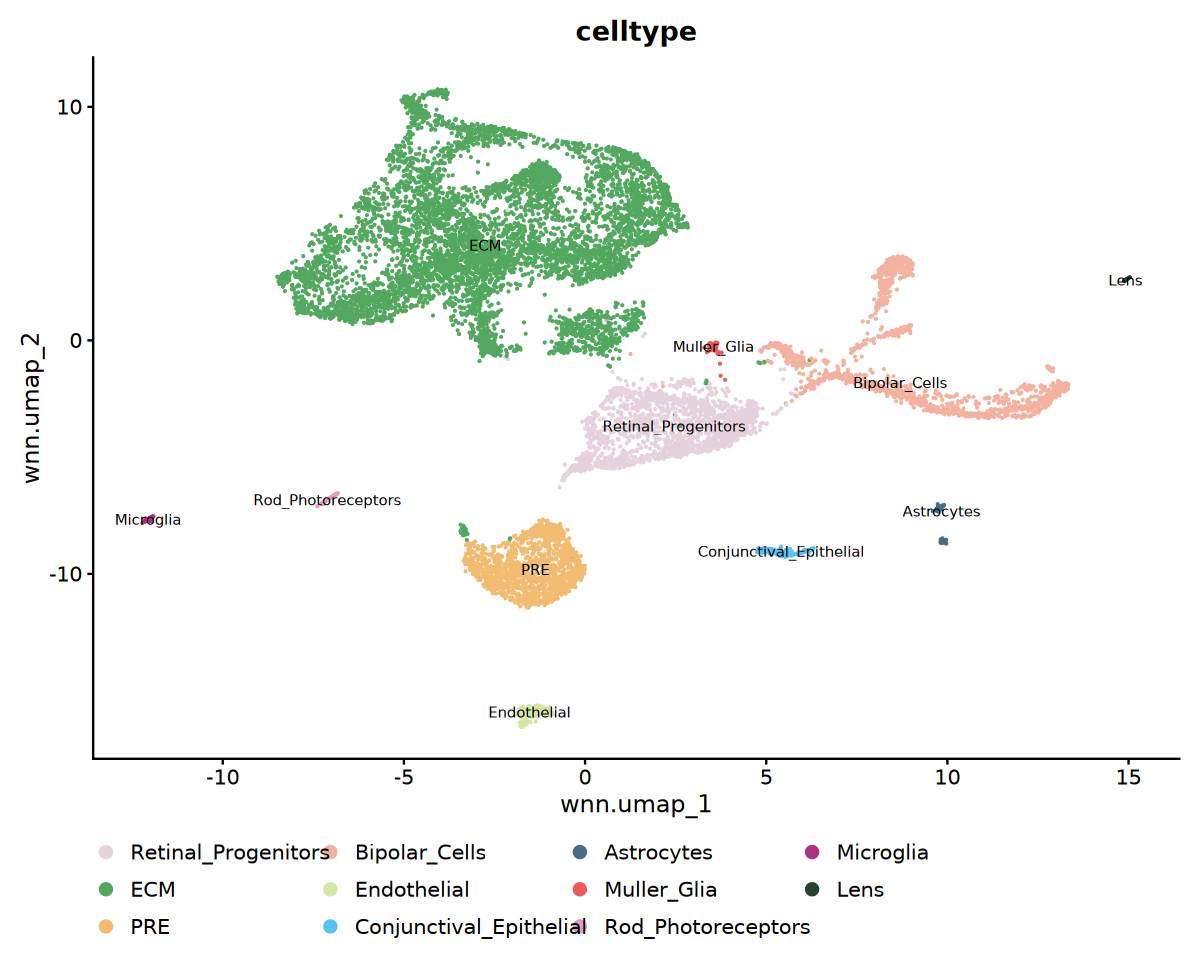
Cell Type Labeling
cat('Starting cell type annotation...', Sys.time(), '\n')
# Cell type annotation based on clustering results (adjust according to actual marker gene expression)
# Here is an example, adjust according to DotPlot results in actual use
celltype_mapping <- c(
"0" = "Retinal_Progenitors",
"1" = "ECM",
"2" = "PRE",
"3" = "ECM",
"4" = "ECM",
"5" = "ECM",
"6" = "Bipolar_Cells",
"7" = "ECM",
"8" = "ECM",
"9" = "ECM",
"10" = "ECM",
"11" = "Bipolar_Cells",
"12" = "Bipolar_Cells",
"13" = "Endothelial",
"14" = "Conjunctival_Epithelial",
"15" = "Astrocytes",
"16" = "Muller_Glia",
"17" = "Rod_Photoreceptors",
"18" = "Microglia",
"19" = "Lens"
)
# Apply cell type annotation
seu$celltype <- recode(
seu$wknn_res.0.5,
!!!celltype_mapping
)Visualization of Annotation Results
cat('Visualizing cell type annotation...', '\n')
# Cell type UMAP visualization
p1 <- DimPlot(
seu,
reduction = "wnn.umap",
group.by = "celltype",
label = TRUE,
label.size = 3,
cols = my36colors
) +
ggtitle("celltype") +
theme(legend.position = "bottom")
# Save cell type annotation plot
pdf("celltype_annotation.pdf", width = 16, height = 12)
print(p1)
dev.off()
options(repr.plot.width=10, repr.plot.height=8)
print(p1)pdf: 2
Save Results
# Save integrated Seurat object
saveRDS(seu, file = "processed.rds")Summary
This tutorial demonstrated the single-sample analysis workflow for single-cell multi-omics data.
Future Analysis Directions
- Cell type annotation and marker gene identification
- Differential expression and differential accessibility analysis
- Co-accessibility analysis of genes and peaks
- Motif analysis
- Footprint analysis
- Gene regulatory network inference
- Trajectory and pseudotime analysis
- CNV analysis
- Joint analysis of epigenetic traits and ATAC
sessionInfo()Platform: x86_64-conda-linux-gnu (64-bit)
Running under: Debian GNU/Linux 12 (bookworm)
Matrix products: default
BLAS/LAPACK: /jp_envs/envs/common/lib/libopenblasp-r0.3.29.so; LAPACK version 3.12.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
time zone: Asia/Shanghai
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] future_1.40.0 harmony_1.2.3
[3] Rcpp_1.0.14 patchwork_1.3.0
[5] ggplot2_3.5.2 dplyr_1.1.4
[7] biovizBase_1.50.0 BSgenome.Hsapiens.UCSC.hg38_1.4.5
[9] BSgenome_1.70.1 rtracklayer_1.62.0
[11] BiocIO_1.12.0 Biostrings_2.70.1
[13] XVector_0.42.0 EnsDb.Hsapiens.v86_2.99.0
[15] ensembldb_2.26.0 AnnotationFilter_1.26.0
[17] GenomicFeatures_1.54.1 AnnotationDbi_1.64.1
[19] Biobase_2.62.0 GenomicRanges_1.54.1
[21] GenomeInfoDb_1.38.1 IRanges_2.36.0
[23] S4Vectors_0.40.2 BiocGenerics_0.48.1
[25] Signac_1.10.0 SeuratObject_4.1.4
[27] Seurat_4.4.0
loaded via a namespace (and not attached):
[1] ProtGenerics_1.34.0 matrixStats_1.5.0
[3] spatstat.sparse_3.1-0 bitops_1.0-9
[5] httr_1.4.7 RColorBrewer_1.1-3
[7] repr_1.1.7 tools_4.3.3
[9] sctransform_0.4.1 backports_1.5.0
[11] R6_2.6.1 lazyeval_0.2.2
[13] uwot_0.2.3 withr_3.0.2
[15] sp_2.2-0 prettyunits_1.2.0
[17] gridExtra_2.3 progressr_0.15.1
[19] cli_3.6.4 Cairo_1.6-2
[21] spatstat.explore_3.4-2 labeling_0.4.3
[23] spatstat.data_3.1-6 ggridges_0.5.6
[25] pbapply_1.7-2 Rsamtools_2.18.0
[27] pbdZMQ_0.3-13 foreign_0.8-87
[29] R.utils_2.13.0 dichromat_2.0-0.1
[31] parallelly_1.43.0 rstudioapi_0.15.0
[33] RSQLite_2.3.9 generics_0.1.3
[35] ica_1.0-3 spatstat.random_3.3-3
[37] Matrix_1.6-5 ggbeeswarm_0.7.2
[39] abind_1.4-5 R.methodsS3_1.8.2
[41] lifecycle_1.0.4 yaml_2.3.10
[43] SummarizedExperiment_1.32.0 SparseArray_1.2.2
[45] BiocFileCache_2.10.1 Rtsne_0.17
[47] grid_4.3.3 blob_1.2.4
[49] promises_1.3.2 crayon_1.5.3
[51] miniUI_0.1.1.1 lattice_0.22-7
[53] cowplot_1.1.3 KEGGREST_1.42.0
[55] pillar_1.10.2 knitr_1.49
[57] rjson_0.2.23 future.apply_1.11.3
[59] codetools_0.2-20 fastmatch_1.1-6
[61] leiden_0.4.3.1 glue_1.8.0
[63] spatstat.univar_3.1-2 data.table_1.17.0
[65] vctrs_0.6.5 png_0.1-8
[67] gtable_0.3.6 cachem_1.1.0
[69] xfun_0.50 S4Arrays_1.2.0
[71] mime_0.13 survival_3.8-3
[73] RcppRoll_0.3.1 fitdistrplus_1.2-2
[75] ROCR_1.0-11 nlme_3.1-168
[77] bit64_4.5.2 progress_1.2.3
[79] filelock_1.0.3 RcppAnnoy_0.0.22
[81] irlba_2.3.5.1 vipor_0.4.7
[83] KernSmooth_2.23-26 rpart_4.1.23
[85] colorspace_2.1-1 DBI_1.2.3
[87] Hmisc_5.2-1 nnet_7.3-19
[89] ggrastr_1.0.2 tidyselect_1.2.1
[91] bit_4.5.0.1 compiler_4.3.3
[93] curl_6.0.1 htmlTable_2.4.3
[95] xml2_1.3.6 DelayedArray_0.28.0
[97] plotly_4.10.4 checkmate_2.3.2
[99] scales_1.3.0 lmtest_0.9-40
[101] rappdirs_0.3.3 stringr_1.5.1
[103] digest_0.6.37 goftest_1.2-3
[105] spatstat.utils_3.1-3 rmarkdown_2.29
[107] htmltools_0.5.8.1 pkgconfig_2.0.3
[109] base64enc_0.1-3 MatrixGenerics_1.14.0
[111] dbplyr_2.5.0 fastmap_1.2.0
[113] rlang_1.1.5 htmlwidgets_1.6.4
[115] shiny_1.10.0 farver_2.1.2
[117] zoo_1.8-14 jsonlite_2.0.0
[119] BiocParallel_1.36.0 R.oo_1.27.0
[121] VariantAnnotation_1.48.1 RCurl_1.98-1.16
[123] magrittr_2.0.3 Formula_1.2-5
[125] GenomeInfoDbData_1.2.11 IRkernel_1.3.2
[127] munsell_0.5.1 reticulate_1.42.0
[129] stringi_1.8.7 zlibbioc_1.48.0
[131] MASS_7.3-60.0.1 plyr_1.8.9
[133] parallel_4.3.3 listenv_0.9.1
[135] ggrepel_0.9.6 deldir_2.0-4
[137] IRdisplay_1.1 splines_4.3.3
[139] tensor_1.5 hms_1.1.3
[141] igraph_2.0.3 uuid_1.2-1
[143] spatstat.geom_3.3-6 reshape2_1.4.4
[145] biomaRt_2.58.0 XML_3.99-0.17
[147] evaluate_1.0.3 httpuv_1.6.15
[149] RANN_2.6.2 tidyr_1.3.1
[151] purrr_1.0.4 polyclip_1.10-7
[153] scattermore_1.2 xtable_1.8-4
[155] restfulr_0.0.15 later_1.4.2
[157] viridisLite_0.4.2 tibble_3.2.1
[159] memoise_2.0.1 beeswarm_0.4.0
[161] GenomicAlignments_1.38.0 cluster_2.1.8.1
[163] globals_0.16.3