SeekArc 单细胞多组学 (RNA+ATAC) 单样本分析教程 (WNN)
环境准备
R 包加载
请选择 common_r 这个环境进行该整合教程的学习
#加载必要的R包
suppressPackageStartupMessages({
library(Seurat)
library(Signac)
library(EnsDb.Hsapiens.v86, lib.loc = "/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/python3.10/lib/R/library")
library(BSgenome.Hsapiens.UCSC.hg38,lib = "/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/python3.10/lib/R/library")
library(biovizBase, lib = "/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/python3.10/lib/R/library")
#library(BSgenome.Mmusculus.UCSC.mm10)
#library(EnsDb.Mmusculus.v79)
library(dplyr)
library(ggplot2)
library(patchwork)
library(harmony)
})
# 设置随机种子
set.seed(1234)
# 设置Seurat选项(注意:8000 * 1024^2 实际上是8GB)
options(future.globals.maxSize = 8000 * 1024^2) # 8GB# 定义颜色方案
my36colors <-c( '#E5D2DD', '#53A85F', '#F1BB72', '#F3B1A0', '#D6E7A3', '#57C3F3', '#476D87',
'#E95C59', '#E59CC4', '#AB3282', '#23452F', '#BD956A', '#8C549C', '#585658',
'#9FA3A8', '#E0D4CA', '#5F3D69', '#C5DEBA', '#58A4C3', '#E4C755', '#F7F398',
'#AA9A59', '#E63863', '#E39A35', '#C1E6F3', '#6778AE', '#91D0BE', '#B53E2B',
'#712820', '#DCC1DD', '#CCE0F5', '#CCC9E6', '#625D9E', '#68A180', '#3A6963',
'#968175', "#6495ED", "#FFC1C1",'#f1ac9d','#f06966','#dee2d1','#6abe83','#39BAE8','#B9EDF8','#221a12',
'#b8d00a','#74828F','#96C0CE','#E95D22','#017890')获取基因注释信息
我们将从 EnsDb 数据库获取相应物种的基因组注释信息(基因位置、转录本、外显子、TSS 等),具体物种需根据数据做更换。这些信息用于:
- 计算 ATAC 的 TSS 富集(判断开放染色质是否在转录起始位点附近更集中)
- 构建基因活性矩阵(把 peaks 信号映射到基因上)
- Peak 注释和功能分析
注意事项:
- 物种与参考基因组版本匹配(如
EnsDb.Hsapiens.v86对应 hg38,EnsDb.Mmusculus.v75对应 mm10) - 染色体命名风格一致(例如都用
chr1、chr2这样的前缀)
# 获取基因注释信息(静默处理警告和消息)
suppressWarnings({
suppressMessages({
annotation <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
seqlevels(annotation) <- paste0('chr', seqlevels(annotation))
genome(annotation) <- 'hg38'
})
})数据读取
本教程提供两种不同形式的数据输入方式,以满足不同用户的数据获取需求,选择适合自己的一种方式即可:
云平台 RDS 文件读取
数据特点:
- RDS 文件是标准的 Seurat 对象文件
- 可直接用于后续的下游分析,也可以提取表达矩阵,重新整合
适用场景:
- 当您无法获得标准的
filtered_feature_bc_matrix表达矩阵时 - 希望利用云平台现有数据进行学习时
- 需要快速重新进行基础分析分析时
注意事项:
- 具体挂载数据和 rds 文件的读取,请参照 jupyter 使用教程
例如下列项目数据/home/demo-SeekGene-com/workspace/data/AY1752565399550/
# 例如下列项目数据/home/demo-seekgene-com/workspace/data/AY1752565399550/
#input <- readRDS("/home/demo-seekgene-com/workspace/data/AY1752565399550/input.rds")
#meta <- read.table("/home/demo-seekgene-com/workspace/data/AY1752565399550/meta.tsv", header=TRUE, sep="\t", row.names = 1)
#提取原有rds数据的基因组注释文件
#annotations <- Annotation(input)
#seu <- CreateSeuratObject(counts = input@assay$RNA@counts, meta.data=meta)
#seu[["ATAC"]] <- CreateChromatinAssay(seu, counts = input@assay$ATAC@counts, fragments = input@assay$ATAC@fragments)
#Annotation(seu) <- annotations
#rm(input)
#gc()标准 filtered_XXXX_bc_matrix 文件读取
适用场景:
- 当您拥有标准的基因表达矩阵和 peaks 开放矩阵文件时
- 希望自主完成单细胞多组学(SeekArc)数据的基础分析时
- 需要进行完整的从原始数据到分析结果的工作流程时
注意:
- 请保证文件结构如下:
数据目录结构需满足如下要求:
- 样本的文件夹名称为样本 ID,如 S127。
- 样本文件夹下包含以下文件:
filtered_feature_bc_matrix:scRNA-seq 表达矩阵文件夹,包含barcodes.tsv.gz、features.tsv.gz和matrix.mtx.gz文件。filtered_peaks_bc_matrix:ATAC 的 peak 开放矩阵文件夹,包含barcodes.tsv.gz、features.tsv.gz和matrix.mtx.gz文件。{样本 ID}_A_fragments.tsv.gz:ATAC 片段文件,如 S127_A_fragments.tsv.gz。{样本 ID}_A_fragments.tsv.gz.tbi:ATAC 片段索引文件,如 S127_A_fragments.tsv.gz.tbi。
具体文件夹结构如下:
├── S127/
│ ├── filtered_feature_bc_matrix/ (scRNA-seq 表达矩阵)
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── filtered_peaks_bc_matrix/ (ATAC 的 peak 开放矩阵)
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── S127_A_fragments.tsv.gz (ATAC 片段文件)
│ └── S127_A_fragments.tsv.gz.tbi
# load the RNA and ATAC data
RNA_counts <- Read10X("./S127/filtered_feature_bc_matrix/")
ATAC_counts <- Read10X("./S127/filtered_peaks_bc_matrix/")
fragpath <- "./S127/S127_A_fragments.tsv.gz"
# create a Seurat object containing the RNA adata
seu <- CreateSeuratObject(
counts = RNA_counts,
assay = "RNA"
)
# create ATAC assay and add it to the object
seu[["ATAC"]] <- CreateChromatinAssay(
counts = ATAC_counts,
sep = c(":", "-"),
fragments = fragpath,
annotation = annotation
)数据质量控制
质控指标计算
RNA 质控指标:
percent.mt: 线粒体基因比例(通常<20%)nfeature_RNA: 总 ATAC 特征数(通常在 200-10000 之间)
ATAC 质控指标:
TSS.enrichment: TSS 富集分数(通常>2)nucleosome_signal: 核小体信号(越低越好)nCount_ATAC: 总 ATAC 计数(通常在 1000-10000 之间)
# 对每个样本进行质量控制
suppressWarnings({
suppressMessages({
# RNA质控指标
seu[["percent.mt"]] <- PercentageFeatureSet(seu, pattern = "^MT-")
# ATAC质控指标
DefaultAssay(seu) <- "ATAC"
# 计算TSS富集分数
seu <- TSSEnrichment(object = seu, fast = FALSE)
# 计算核小体信号
seu <- NucleosomeSignal(object = seu)
})
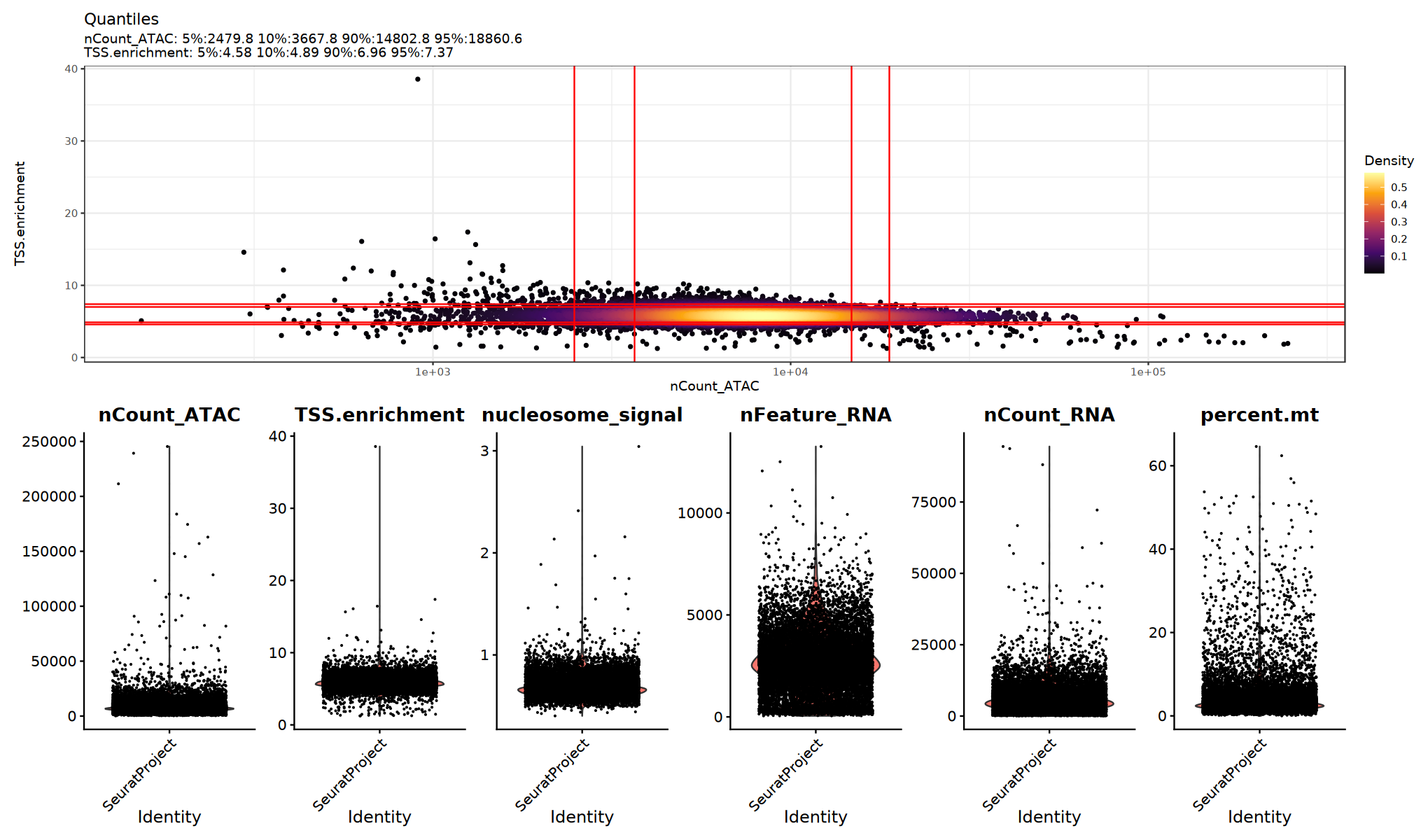
})质控指标可视化
用小提琴图查看各 QC 指标的分布/异常点,确定合适的阈值:
建议:
- 观察是否存在明显的长尾或双峰分布
- 尝试多组阈值并比较下游聚类/UMAP 是否更清晰
# 可视化各质控指标,该cell内容选择性执行,非必要
options(repr.plot.width = 17, repr.plot.height = 10)
suppressWarnings({
p1=DensityScatter(seu, x = 'nCount_ATAC', y = 'TSS.enrichment', log_x = TRUE, quantiles = TRUE)
p2=VlnPlot(
object = seu,
features = c('nCount_ATAC', 'TSS.enrichment', 'nucleosome_signal',"nFeature_RNA", "nCount_RNA", "percent.mt"),
pt.size = 0.1,
ncol = 6
)
print(p1 / p2)
})
低质量细胞过滤
根据质控指标过滤低质量细胞。具体阈值应根据数据特征进行调整。具体过滤阈值参考上面的小提琴图分布情况。
# 质控过滤
cells_before <- ncol(seu)
seu <- subset(seu,
subset = nFeature_RNA > 200 &
nFeature_RNA < 8000 &
nCount_RNA > 500 &
nCount_RNA < 30000 &
percent.mt < 20 &
nCount_ATAC > 500 &
nCount_ATAC < 100000 &
TSS.enrichment > 1 &
nucleosome_signal < 2
)
cells_after <- ncol(seu)数据标准化处理
RNA 数据标准化和线性降维
标准化方法: 默认情况下,Seurat 采用"LogNormalize"全局缩放标准化方法:
- 首先根据总表达量对每个细胞的特征表达量进行标准化
- 乘以比例因子(默认为 10,000)
- 然后对结果进行对数转换
特征选择: 计算数据集中呈现高细胞间变异性的特征子集(即某些细胞中高表达,而其他细胞中低表达的特征)
数据缩放: 执行线性变换("scaling"),这是主成分分析(PCA)等降维技术前的标准预处理步骤
降维分析: 对标准化后的数据执行主成分分析(PCA)
suppressWarnings({
suppressMessages({
DefaultAssay(seu) <- "RNA"
seu <- NormalizeData(seu, assay = "RNA")
seu <- FindVariableFeatures(seu, assay = "RNA", selection.method = "vst", nfeatures = 2000)
seu <- ScaleData(seu, assay = "RNA")
seu <- RunPCA(seu, assay = "RNA", npcs = 50)
})
})ATAC 数据标准化和线性降维
标准化方法: Signac 采用词频-逆文档频率(TF-IDF)标准化方法:
- 在细胞间进行标准化以校正测序深度差异
- 在峰区间进行标准化使稀有峰获得更高权重
特征选择策略: 由于单细胞 ATAC 测序数据的动态范围较低,我们无法像处理单细胞 RNA 测序数据那样直接进行可变特征选择。作为替代方案,可通过 FindTopFeatures()函数选择仅使用前 n%的特征(峰),或剔除在少于 n 个细胞中出现的特征。
降维分析: 随后使用上述筛选的特征(峰)对 TF-IDF 矩阵进行奇异值分解(SVD)。
suppressWarnings({
suppressMessages({
seu <- RunTFIDF(seu, assay = "ATAC")
seu <- FindTopFeatures(seu, assay = "ATAC", min.cutoff = 'q0')
seu <- RunSVD(seu, assay = "ATAC")
})
})非线性降维与聚类
三种聚类策略
- RNA 聚类:基于基因表达相似性
- ATAC 聚类:基于染色质可及性相似性
- WNN 聚类:整合两种模态信息(推荐)
结果解读
- 不同方法可能产生不同的聚类结果
- WNN 聚类通常能发现更细致的细胞亚群
- 建议优先使用 WNN 结果进行下游分析
RNA 降维与聚类
RNA 聚类策略:
- 基于 RNA 数据的独立聚类分析
- 利用基因表达模式进行细胞分类
suppressWarnings({
suppressMessages({
seu <- RunUMAP(seu, reduction = "pca", dims = 1:30, assay = "RNA",reduction.name="rnaumap")
seu <- FindNeighbors(seu, reduction = "pca", dims = 1:30, assay = "RNA",graph.name = "rnaneigobr")
seu <- FindClusters(seu, resolution = 0.5, algorithm = 1,graph.name = "rnaneigobr")
})
})Number of nodes: 12694
Number of edges: 119839
Running Louvain algorithm...n Maximum modularity in 10 random starts: 0.9108
Number of communities: 25
Elapsed time: 0 seconds
ATAC 降维与聚类
ATAC 聚类策略:
- 基于 ATAC 数据的独立聚类分析
- 利用染色质可及性模式进行细胞分类
suppressWarnings({
suppressMessages({
seu <- RunUMAP(seu, reduction = "lsi", dims = 1:30, assay = "ATAC",reduction.name="atacumap")
seu <- FindNeighbors(seu, reduction = "lsi", dims = 1:30, assay = "ATAC",graph.name = "atacneigobr")
seu <- FindClusters(seu, resolution = 0.5, algorithm = 1,graph.name = "atacneigobr")
})
})Number of nodes: 12694
Number of edges: 120530
Running Louvain algorithm...n Maximum modularity in 10 random starts: 0.8927
Number of communities: 19
Elapsed time: 0 seconds
加权最近邻(WNN)分析
WNN 整合聚类策略:
- 结合 RNA 和 ATAC 数据的加权最近邻分析
- 提供更准确的细胞类型识别
suppressWarnings({
suppressMessages({
seu <- FindMultiModalNeighbors(seu, reduction.list = list("pca", "lsi"), dims.list = list(1:30, 1:30))
# 基于WNN进行聚类
seu <- FindClusters(seu, graph.name = "wknn", resolution = 0.5)
# WNN UMAP
seu <- RunUMAP(seu, nn.name = "weighted.nn", reduction.name = "wnn.umap")
})
})Number of nodes: 12694
Number of edges: 191058
Running Louvain algorithm...n Maximum modularity in 10 random starts: 0.8993
Number of communities: 20
Elapsed time: 0 seconds
可视化
# 比较不同方法的聚类结果
p1 <- DimPlot(seu, reduction = "rnaumap", group.by = "rnaneigobr_res.0.5",label=T, cols = my36colors) + ggtitle("RNA")
p2 <- DimPlot(seu, reduction = "atacumap", group.by = "atacneigobr_res.0.5",label=T, cols = my36colors) + ggtitle("ATAC")
p3 <- DimPlot(seu, reduction = "wnn.umap", group.by = "wknn_res.0.5",label=T, cols = my36colors) + ggtitle("WNN")
options(repr.plot.width=23, repr.plot.height=6)
print(p1 + p2 + p3)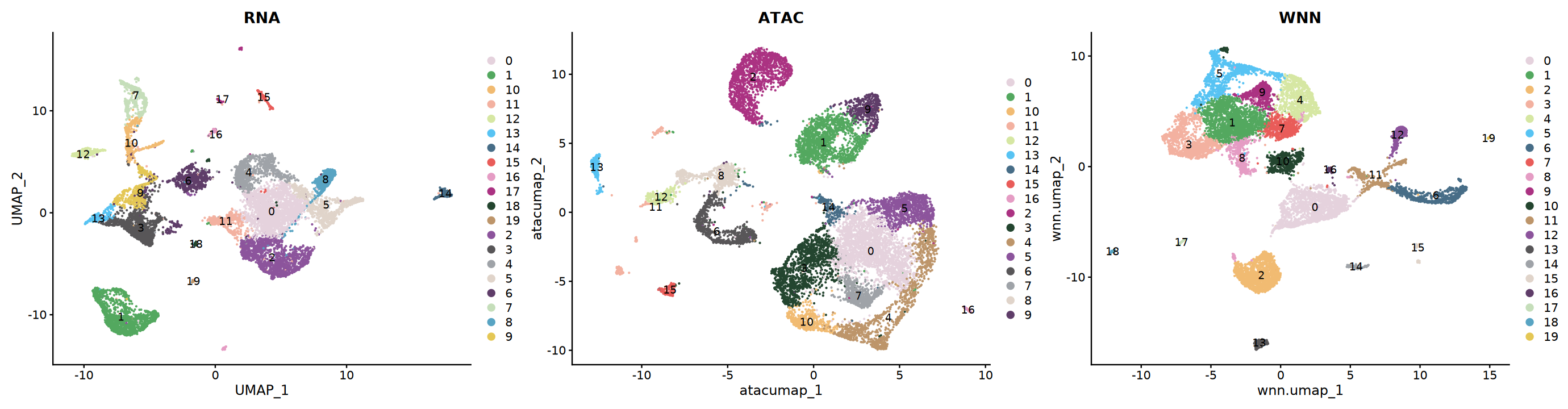
细胞类型注释
marker 基因
根据组织类型,收集不同细胞类型 marker 基因集,本示例数据是眼睛,下面是眼睛中细胞类型及其对应的 marker 基因,用气泡图可视化不同 cluster 高表达哪些细胞类型的 marker 基因
eye_marker_integrated <- list(
# ========== 光感受器细胞 ==========
"Rod_Photoreceptors" = c("RHO", "PDE6B", "CNGB1", "PDE6A", "NR2E3", "REEP6",
"RCVRN", "SAG", "NEB", "SLC24A3", "TRPM1"),
"Cone_Photoreceptors" = c("ARR3", "GNAT2", "OPN1SW", "TRPM3"),
# ========== 视网膜神经元 ==========
"Bipolar_Cells" = c("VSX1", "OTX2", "GRM6", "PRKCA",
"DLG2", "NRXN3", "RBFOX3", "FSTL4"),
"Amacrine_Cells" = c("GAD1", "GAD2", "C1QL2", "TFAP2B", "SLC32A1"),
"Horizontal_Cells" = c("ONECUT1", "LHX1", "CALB1", "NFIA"),
"Retinal_Ganglion_Cells" = c("RBPMS", "THY1", "NEFL", "POU4F2"),
# ========== 胶质细胞 ==========
"Muller_Glia" = c("SLC1A3", "RLBP1", "SOX9", "CRYAB"),
"Astrocytes" = c("GFAP", "AQP4", "S100B"),
"Microglia" = c("TMEM119", "C1QA", "AIF1", "CX3CR1", "CD74",
"PTPRC", "CSF1R", "CCL3L1", "SPP1", "P2RY12"),
# ========== 血管与支持细胞 ==========
"Endothelial" = c("FLT1", "PODXL", "PLVAP", "KDR", "EGFL7",
"CLDN5", "PECAM1", "CDH5"),
"Pericytes" = c("PDGFRB", "RGS5", "CSPG4", "STAB1"),
# ========== 上皮细胞 ==========
"RPE" = c("RPE65", "BEST1", "PMEL", "TYRP1", "DCT", "SLC38A11"),
#"Corneal_Epithelial" = c("KRT12", "KRT3"),
"Corneal_Endothelial" = c("SLC4A11", "COL8A2", "ATP1A1"),
"Conjunctival_Epithelial" = c("KRT13", "KRT19", "MUC5AC"),
"Retinal_Progenitors" = c("VSX2", "PAX6","SOX2", "HES1", "NOTCH1"), # 神经视网膜增殖期
# ========== 晶状体细胞 ==========
"Lens_Epithelial" = c("CRYAA", "BFSP1", "MIP"),
"Lens_Fiber" = c("CRYGS", "LIM2", "FN1"),
# ========== 其他细胞 ==========
"Melanocytes" = c("TYR", "MLANA", "MITF"),
#"Erythrocytes" = c("HBB", "HBA1", "HBA2"),
"ECM" = c("COL1A1", "COL3A1", "COL12A1", "COL1A2", "COL6A3")#,
#"Others" = c("TTN", "CLCN5", "DCC", "MIAT")
)# 设置图形大小
options(repr.plot.width=25, repr.plot.height=7)
# 绘制DotPlot
DefaultAssay(seu)="RNA"
DotPlot(seu,
group.by = "wknn_res.0.5",
features = eye_marker_integrated,
cols = c("#f8f8f8","#ff3472"),
#dot.min = 0.05,
dot.scale = 8)+ # 应用自定义配色
RotatedAxis() +
scale_x_discrete("") +
scale_y_discrete("") +
theme(
axis.text.x = element_text(size = 12, face = "bold",
angle = 45, hjust = 1, vjust = 1),
axis.text.y = element_text(size = 12, face = "bold"),
plot.title = element_text(size = 14, face = "bold", hjust = 0.5),
legend.title = element_text(size = 10, face = "bold")
) +
ggtitle("Marker Genes Expression") +
labs(color = "Expression\nLevel") # 修改图例标题"The \`facets\` argument of \`facet_grid()\` is deprecated as of ggplot2 2.2.0.
ℹ Please use the \`rows\` argument instead.
ℹ The deprecated feature was likely used in the Seurat package.
Please report the issue at
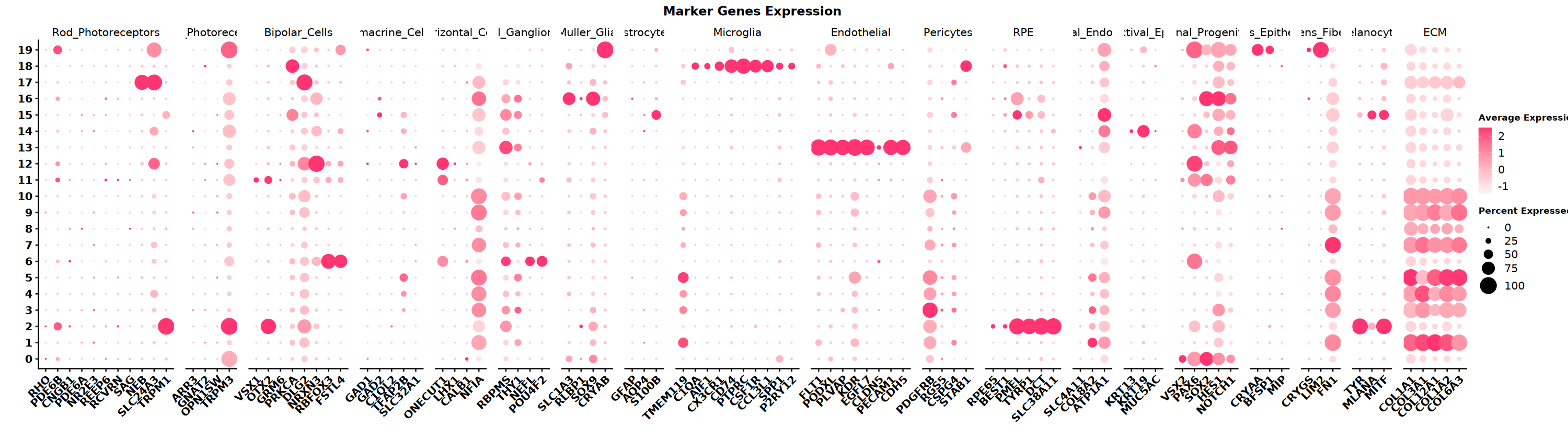
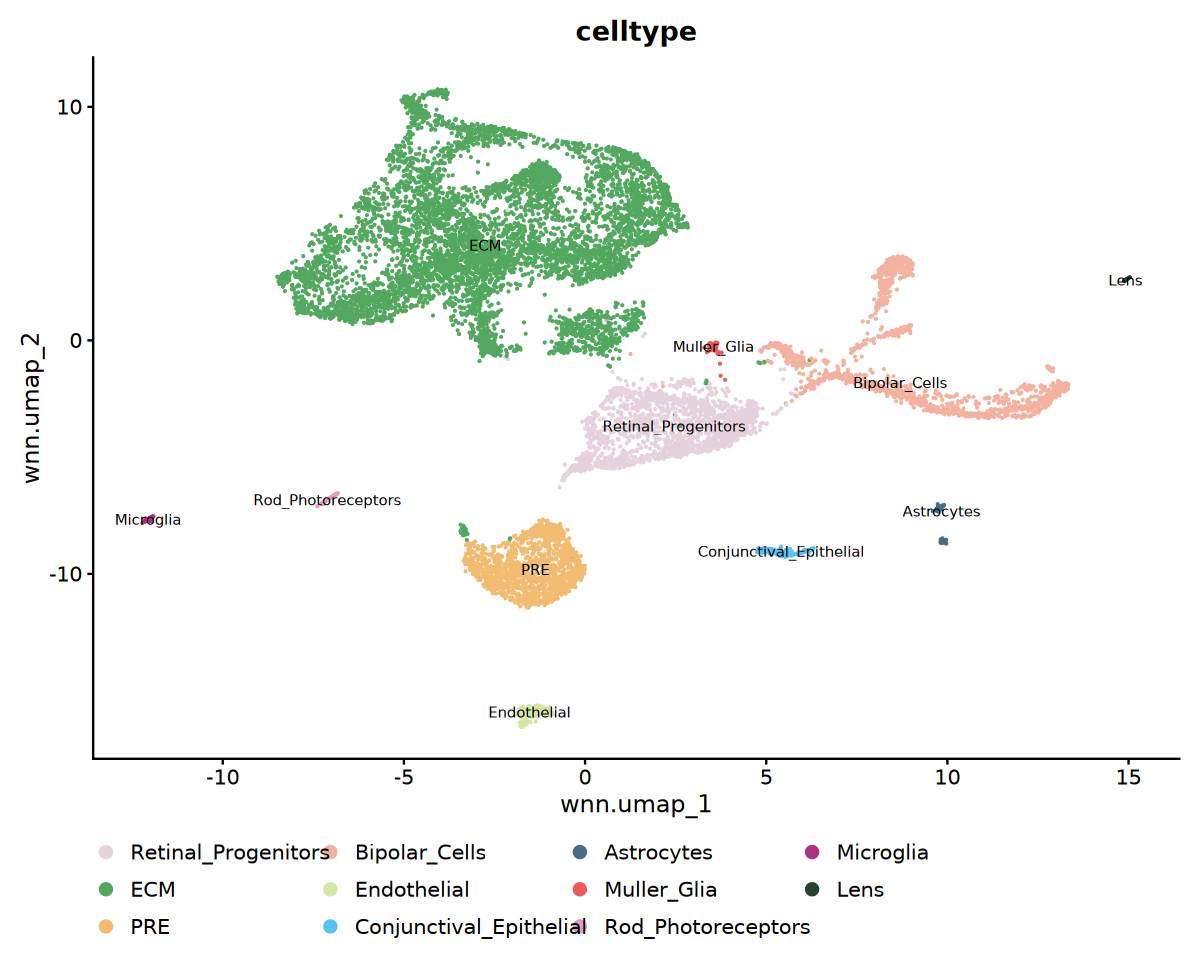
** 注意事项 ** SeekArc 单细胞双组学数据细胞注释一般基于 wnn 多模态降维,依据 marker 基因在 wknn 聚类结果中表达情况进行注释
options(repr.plot.width=10, repr.plot.height=8)
DimPlot(seu, reduction = "wnn.umap", group.by = "wknn_res.0.5", cols = my36colors,label = T)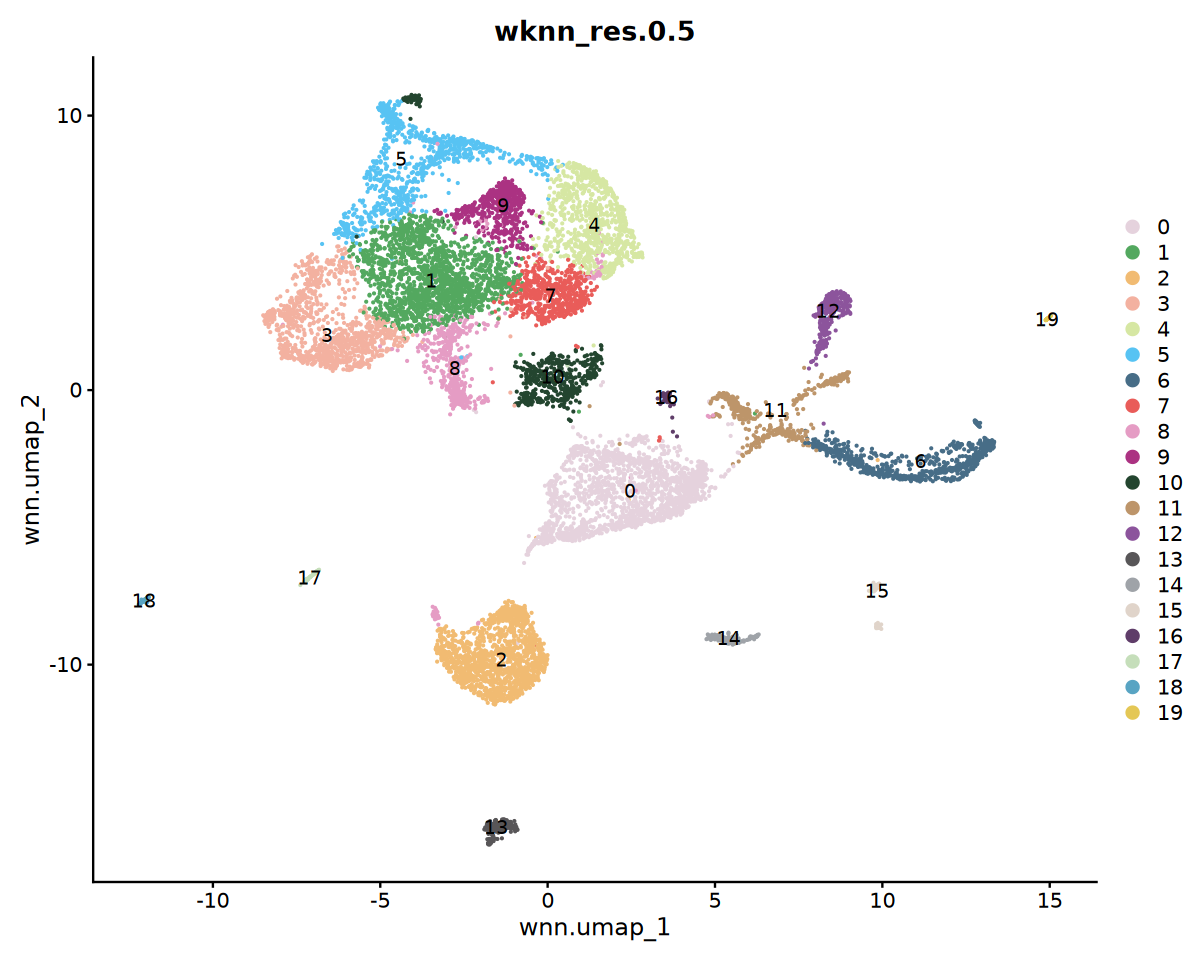
细胞类型标注
cat('开始细胞类型注释...', Sys.time(), '\n')
# 基于聚类结果进行细胞类型注释(需要根据实际的marker基因表达情况调整)
# 这里提供一个示例,实际使用时需要根据DotPlot结果进行调整
celltype_mapping <- c(
"0" = "Retinal_Progenitors",
"1" = "ECM",
"2" = "PRE",
"3" = "ECM",
"4" = "ECM",
"5" = "ECM",
"6" = "Bipolar_Cells",
"7" = "ECM",
"8" = "ECM",
"9" = "ECM",
"10" = "ECM",
"11" = "Bipolar_Cells",
"12" = "Bipolar_Cells",
"13" = "Endothelial",
"14" = "Conjunctival_Epithelial",
"15" = "Astrocytes",
"16" = "Muller_Glia",
"17" = "Rod_Photoreceptors",
"18" = "Microglia",
"19" = "Lens"
)
# 应用细胞类型注释
seu$celltype <- recode(
seu$wknn_res.0.5,
!!!celltype_mapping
)注释结果可视化
cat('细胞类型注释可视化...', '\n')
# 细胞类型UMAP可视化
p1 <- DimPlot(
seu,
reduction = "wnn.umap",
group.by = "celltype",
label = TRUE,
label.size = 3,
cols = my36colors
) +
ggtitle("celltype") +
theme(legend.position = "bottom")
# 保存细胞类型注释图
pdf("celltype_annotation.pdf", width = 16, height = 12)
print(p1)
dev.off()
options(repr.plot.width=10, repr.plot.height=8)
print(p1)pdf: 2
保存结果
# 保存整合后的Seurat对象
saveRDS(seu, file = "processed.rds")总结
本教程演示了单细胞多组学数据的单样本分析流程。
后续分析方向
- 细胞类型注释和标记基因识别
- 差异表达和差异可及性分析
- genes 和 peaks 的共可及性分析
- Motif 分析
- 足迹分析
- 基因调控网络推断
- 发育轨迹和伪时间分析
- 拷贝数变异分析
- 表观性状与 ATAC 联合分析
sessionInfo()Platform: x86_64-conda-linux-gnu (64-bit)
Running under: Debian GNU/Linux 12 (bookworm)
Matrix products: default
BLAS/LAPACK: /jp_envs/envs/common/lib/libopenblasp-r0.3.29.so; LAPACK version 3.12.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
time zone: Asia/Shanghai
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] future_1.40.0 harmony_1.2.3
[3] Rcpp_1.0.14 patchwork_1.3.0
[5] ggplot2_3.5.2 dplyr_1.1.4
[7] biovizBase_1.50.0 BSgenome.Hsapiens.UCSC.hg38_1.4.5
[9] BSgenome_1.70.1 rtracklayer_1.62.0
[11] BiocIO_1.12.0 Biostrings_2.70.1
[13] XVector_0.42.0 EnsDb.Hsapiens.v86_2.99.0
[15] ensembldb_2.26.0 AnnotationFilter_1.26.0
[17] GenomicFeatures_1.54.1 AnnotationDbi_1.64.1
[19] Biobase_2.62.0 GenomicRanges_1.54.1
[21] GenomeInfoDb_1.38.1 IRanges_2.36.0
[23] S4Vectors_0.40.2 BiocGenerics_0.48.1
[25] Signac_1.10.0 SeuratObject_4.1.4
[27] Seurat_4.4.0
loaded via a namespace (and not attached):
[1] ProtGenerics_1.34.0 matrixStats_1.5.0
[3] spatstat.sparse_3.1-0 bitops_1.0-9
[5] httr_1.4.7 RColorBrewer_1.1-3
[7] repr_1.1.7 tools_4.3.3
[9] sctransform_0.4.1 backports_1.5.0
[11] R6_2.6.1 lazyeval_0.2.2
[13] uwot_0.2.3 withr_3.0.2
[15] sp_2.2-0 prettyunits_1.2.0
[17] gridExtra_2.3 progressr_0.15.1
[19] cli_3.6.4 Cairo_1.6-2
[21] spatstat.explore_3.4-2 labeling_0.4.3
[23] spatstat.data_3.1-6 ggridges_0.5.6
[25] pbapply_1.7-2 Rsamtools_2.18.0
[27] pbdZMQ_0.3-13 foreign_0.8-87
[29] R.utils_2.13.0 dichromat_2.0-0.1
[31] parallelly_1.43.0 rstudioapi_0.15.0
[33] RSQLite_2.3.9 generics_0.1.3
[35] ica_1.0-3 spatstat.random_3.3-3
[37] Matrix_1.6-5 ggbeeswarm_0.7.2
[39] abind_1.4-5 R.methodsS3_1.8.2
[41] lifecycle_1.0.4 yaml_2.3.10
[43] SummarizedExperiment_1.32.0 SparseArray_1.2.2
[45] BiocFileCache_2.10.1 Rtsne_0.17
[47] grid_4.3.3 blob_1.2.4
[49] promises_1.3.2 crayon_1.5.3
[51] miniUI_0.1.1.1 lattice_0.22-7
[53] cowplot_1.1.3 KEGGREST_1.42.0
[55] pillar_1.10.2 knitr_1.49
[57] rjson_0.2.23 future.apply_1.11.3
[59] codetools_0.2-20 fastmatch_1.1-6
[61] leiden_0.4.3.1 glue_1.8.0
[63] spatstat.univar_3.1-2 data.table_1.17.0
[65] vctrs_0.6.5 png_0.1-8
[67] gtable_0.3.6 cachem_1.1.0
[69] xfun_0.50 S4Arrays_1.2.0
[71] mime_0.13 survival_3.8-3
[73] RcppRoll_0.3.1 fitdistrplus_1.2-2
[75] ROCR_1.0-11 nlme_3.1-168
[77] bit64_4.5.2 progress_1.2.3
[79] filelock_1.0.3 RcppAnnoy_0.0.22
[81] irlba_2.3.5.1 vipor_0.4.7
[83] KernSmooth_2.23-26 rpart_4.1.23
[85] colorspace_2.1-1 DBI_1.2.3
[87] Hmisc_5.2-1 nnet_7.3-19
[89] ggrastr_1.0.2 tidyselect_1.2.1
[91] bit_4.5.0.1 compiler_4.3.3
[93] curl_6.0.1 htmlTable_2.4.3
[95] xml2_1.3.6 DelayedArray_0.28.0
[97] plotly_4.10.4 checkmate_2.3.2
[99] scales_1.3.0 lmtest_0.9-40
[101] rappdirs_0.3.3 stringr_1.5.1
[103] digest_0.6.37 goftest_1.2-3
[105] spatstat.utils_3.1-3 rmarkdown_2.29
[107] htmltools_0.5.8.1 pkgconfig_2.0.3
[109] base64enc_0.1-3 MatrixGenerics_1.14.0
[111] dbplyr_2.5.0 fastmap_1.2.0
[113] rlang_1.1.5 htmlwidgets_1.6.4
[115] shiny_1.10.0 farver_2.1.2
[117] zoo_1.8-14 jsonlite_2.0.0
[119] BiocParallel_1.36.0 R.oo_1.27.0
[121] VariantAnnotation_1.48.1 RCurl_1.98-1.16
[123] magrittr_2.0.3 Formula_1.2-5
[125] GenomeInfoDbData_1.2.11 IRkernel_1.3.2
[127] munsell_0.5.1 reticulate_1.42.0
[129] stringi_1.8.7 zlibbioc_1.48.0
[131] MASS_7.3-60.0.1 plyr_1.8.9
[133] parallel_4.3.3 listenv_0.9.1
[135] ggrepel_0.9.6 deldir_2.0-4
[137] IRdisplay_1.1 splines_4.3.3
[139] tensor_1.5 hms_1.1.3
[141] igraph_2.0.3 uuid_1.2-1
[143] spatstat.geom_3.3-6 reshape2_1.4.4
[145] biomaRt_2.58.0 XML_3.99-0.17
[147] evaluate_1.0.3 httpuv_1.6.15
[149] RANN_2.6.2 tidyr_1.3.1
[151] purrr_1.0.4 polyclip_1.10-7
[153] scattermore_1.2 xtable_1.8-4
[155] restfulr_0.0.15 later_1.4.2
[157] viridisLite_0.4.2 tibble_3.2.1
[159] memoise_2.0.1 beeswarm_0.4.0
[161] GenomicAlignments_1.38.0 cluster_2.1.8.1
[163] globals_0.16.3