SeekSpace Advanced Analysis: Neighborhood Enrichment and Co-occurrence Analysis Based on Squidpy
Select execution environment 'cellcharter-env'
Squidpy is a tool for analyzing and visualizing spatial data. Built on top of scanpy and anndata, it inherits modularity and scalability. It provides analysis tools utilizing spatial coordinates of data and tissue images (if available).
Analysis content mainly includes:
- Neighborhood Enrichment
- Co-occurrence Analysis
- Variable Genes
- Dispersion Level
Data Loading and Preprocessing
Read based on three matrix files, additionally adding spatial location and cell annotation (can be any cell clustering) results.
adata = sc.read_10x_mtx(
"/PROJ2/FLOAT/weichendan/seekspace/WTH1092/demo_WTH1092/Outs/WTH1092_filtered_feature_bc_matrix/",
cache=True
)adatavar: 'gene_ids', 'feature_types'
adata.var_names_make_unique()
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
adata.var["mt"] = adata.var_names.str.startswith("mt-")
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :].copy()
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
adata.raw = adata
adata = adata[:, adata.var.highly_variable]
sc.pp.regress_out(adata, ["total_counts", "pct_counts_mt"])
sc.pp.scale(adata, max_value=10)
sc.tl.pca(adata, svd_solver="arpack")
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
sc.tl.umap(adata)
sc.tl.tsne(adata)df[key] = c
/PROJ2/FLOAT/weichendan/software/micromamba/envs/cellcharter-env/lib/python3.9/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
adataobs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'feature_types', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne'
obsm: 'X_pca', 'X_umap', 'X_tsne'
varm: 'PCs'
obsp: 'distances', 'connectivities'
spatial_df = pd.read_csv("/PROJ2/FLOAT/weichendan/seekspace/WTH1092/demo_WTH1092/Outs/WTH1092_filtered_feature_bc_matrix/cell_locations.tsv.gz",index_col=0,sep='\t')
spatial_df.columns = ["spatial_1","spatial_2"]
spatial_df = pd.merge(adata.obs, spatial_df, left_index=True, right_index=True)[["spatial_1","spatial_2"]]
adata.obsm["spatial"] = spatial_df.valuesspatial_df| spatial_1 | spatial_2 | |
|---|---|---|
| TTGTGTTATGCAAGAGGTAGTCATACG | 33195 | 11954 |
| TGCGACGATGACGATACCTAAGTACGA | 25664 | 15747 |
| ACCTGAACGAAGGGTCAGCTCTGCTTC | 36614 | 13290 |
| CACTGGGTACGGTCGACCCTAATAACG | 35019 | 10923 |
| AAGATAGCGAAGGAGGGCTAAGTACGA | 42849 | 13006 |
| ... | ... | ... |
| TCAAGCAGCTGTTACCATTCTGCGCTC | 17118 | 14572 |
| ACCCTTGCGAACTCAACGCTCTGCTTC | 34011 | 13369 |
| GTTCTATATGCGGACTGGCTCTGCTTC | 15772 | 12816 |
| TACCGAATACCTCCGACGCTCTGCTTC | 21987 | 16150 |
| AATGAAGGCTCTTGAACAGCGACGGAT | 29200 | 4941 |
22512 rows × 2 columns
cell_anno = pd.read_csv("/PROJ2/FLOAT/weichendan/seekspace/WTH1092/annotation.csv",index_col=0)
cell_anno = cell_anno[["Main_CellType","Sub_CellType"]]
adata.obs = pd.merge(adata.obs,cell_anno,left_index=True,right_index=True)adataobs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'Main_CellType', 'Sub_CellType'
var: 'gene_ids', 'feature_types', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne'
obsm: 'X_pca', 'X_umap', 'X_tsne', 'spatial'
varm: 'PCs'
obsp: 'distances', 'connectivities'
Major Group Annotation Results:
sc.settings.set_figure_params(figsize=(10, 5))
sc.pl.embedding(adata,"spatial",color=["Main_CellType"])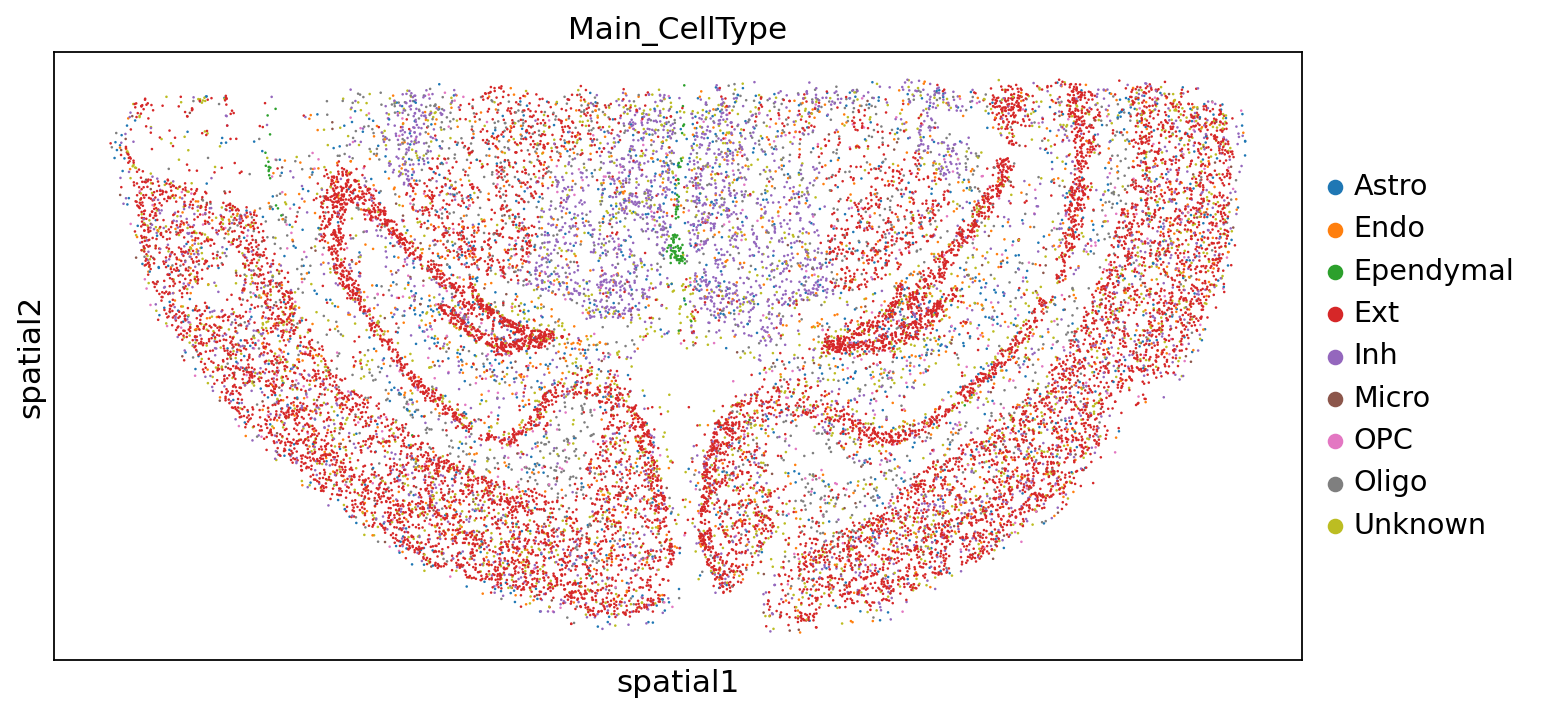
Subgroup Annotation Results:
sc.settings.set_figure_params(figsize=(10, 5))
sc.pl.embedding(adata,"spatial",color=["Sub_CellType"])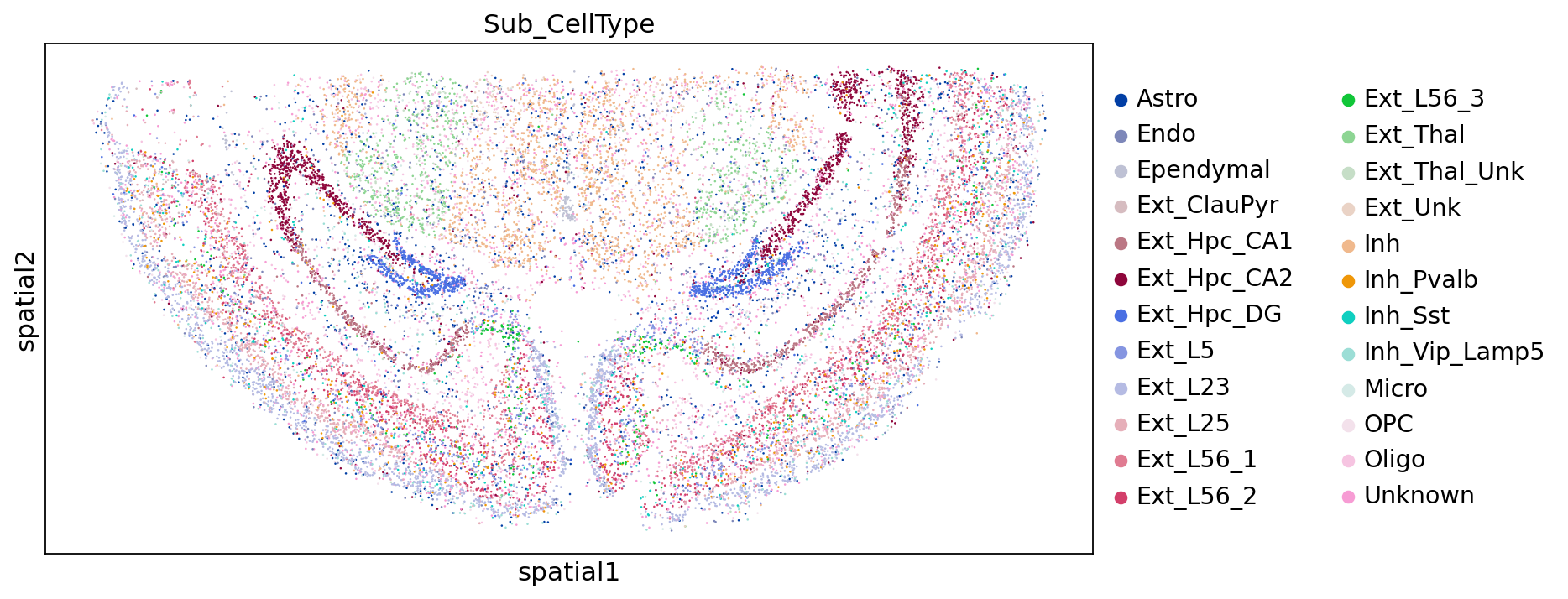
Neighborhood Enrichment
Calculating neighborhood enrichment helps us identify cell clusters sharing common neighborhood structures across the tissue.
In short, this is an enrichment score for cluster spatial proximity: if cells belonging to two different clusters are usually close to each other, they will have a high score and can be defined as enriched. On the other hand, if they are far apart and rarely neighbors, the score will be low and can be defined as depleted.
sq.gr.spatial_neighbors(adata)
sq.gr.nhood_enrichment(adata, cluster_key="Sub_CellType")adataobs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'Main_CellType', 'Sub_CellType'
var: 'gene_ids', 'feature_types', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne', 'Main_CellType_colors', 'Sub_CellType_colors', 'spatial_neighbors', 'Sub_CellType_nhood_enrichment'
obsm: 'X_pca', 'X_umap', 'X_tsne', 'spatial'
varm: 'PCs'
obsp: 'distances', 'connectivities', 'spatial_connectivities', 'spatial_distances'
sq.pl.nhood_enrichment(adata, cluster_key="Sub_CellType", figsize=(5, 10))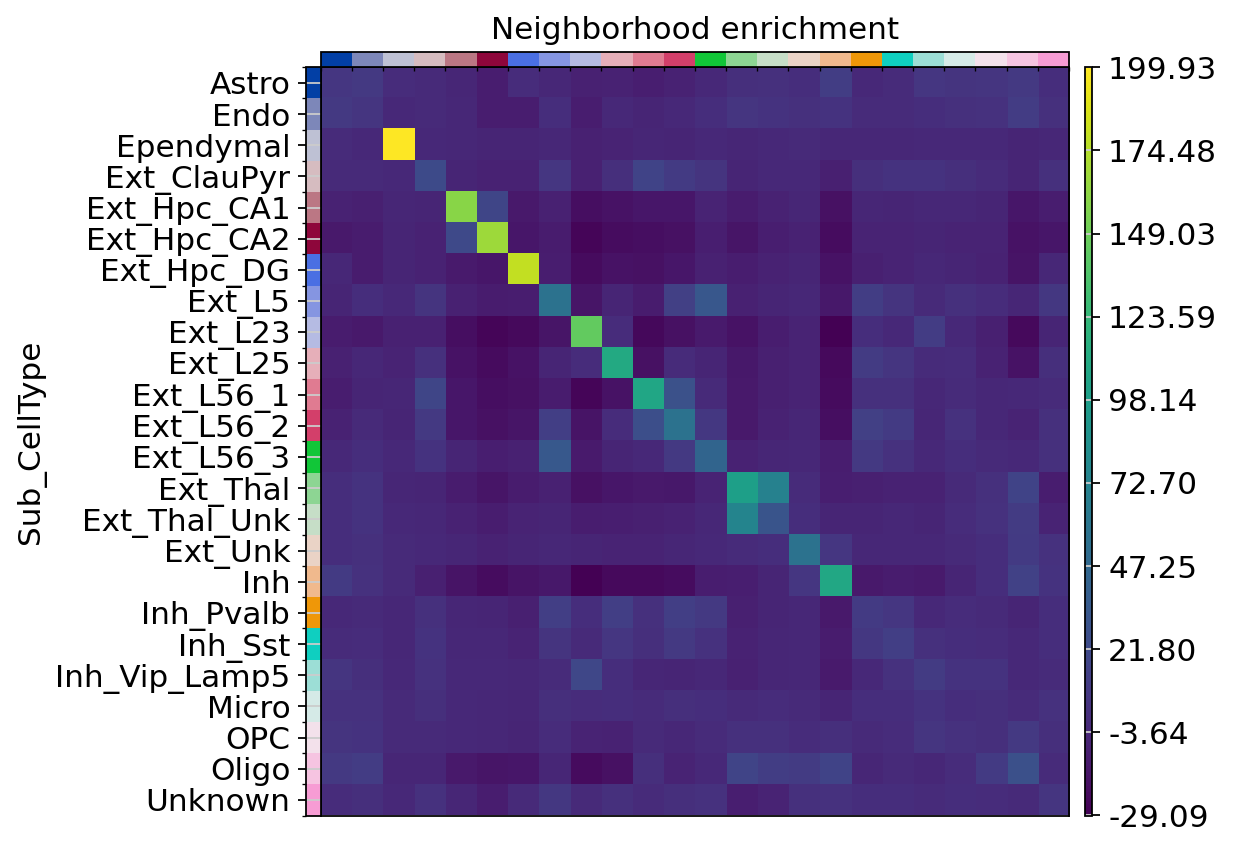
Figure Legend: Neighborhood enrichment analysis between cell clusters in spatial coordinates.
Spatial Dimension Co-occurrence Analysis
Besides neighborhood enrichment scores, we can visualize cluster co-occurrence in the spatial dimension. This is similar to the analysis above but runs on raw spatial coordinates rather than the data matrix.
sq.gr.co_occurrence(adata, cluster_key="Sub_CellType")adataobs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'Main_CellType', 'Sub_CellType'
var: 'gene_ids', 'feature_types', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne', 'Main_CellType_colors', 'Sub_CellType_colors', 'spatial_neighbors', 'Sub_CellType_nhood_enrichment', 'Sub_CellType_co_occurrence'
obsm: 'X_pca', 'X_umap', 'X_tsne', 'spatial'
varm: 'PCs'
obsp: 'distances', 'connectivities', 'spatial_connectivities', 'spatial_distances'
np.unique(adata.obs['Sub_CellType'])'Ext_Hpc_CA2', 'Ext_Hpc_DG', 'Ext_L23', 'Ext_L25', 'Ext_L5',
'Ext_L56_1', 'Ext_L56_2', 'Ext_L56_3', 'Ext_Thal', 'Ext_Thal_Unk',
'Ext_Unk', 'Inh', 'Inh_Pvalb', 'Inh_Sst', 'Inh_Vip_Lamp5', 'Micro',
'OPC', 'Oligo', 'Unknown'], dtype=object)
import matplotlib.pyplot as plt
sq.pl.co_occurrence(
adata,
cluster_key="Sub_CellType",
clusters="Astro",
figsize=(10, 4)
)
# Get legend on current axes (if any)
legend = plt.gca().get_legend()
if legend is not None:
# Get legend handles and labels
handles, labels = legend.legend_handles, [t.get_text() for t in legend.texts]
# Remove old legend
legend.remove()
# Add new legend and set ncol=2
plt.legend(
handles=handles,
labels=labels,
loc='center left', # Or specify position, e.g., 'upper right'
bbox_to_anchor=(1, 0.5), # Place to the right of axes (1 means outside right boundary)
ncol=2, # Set to two columns
)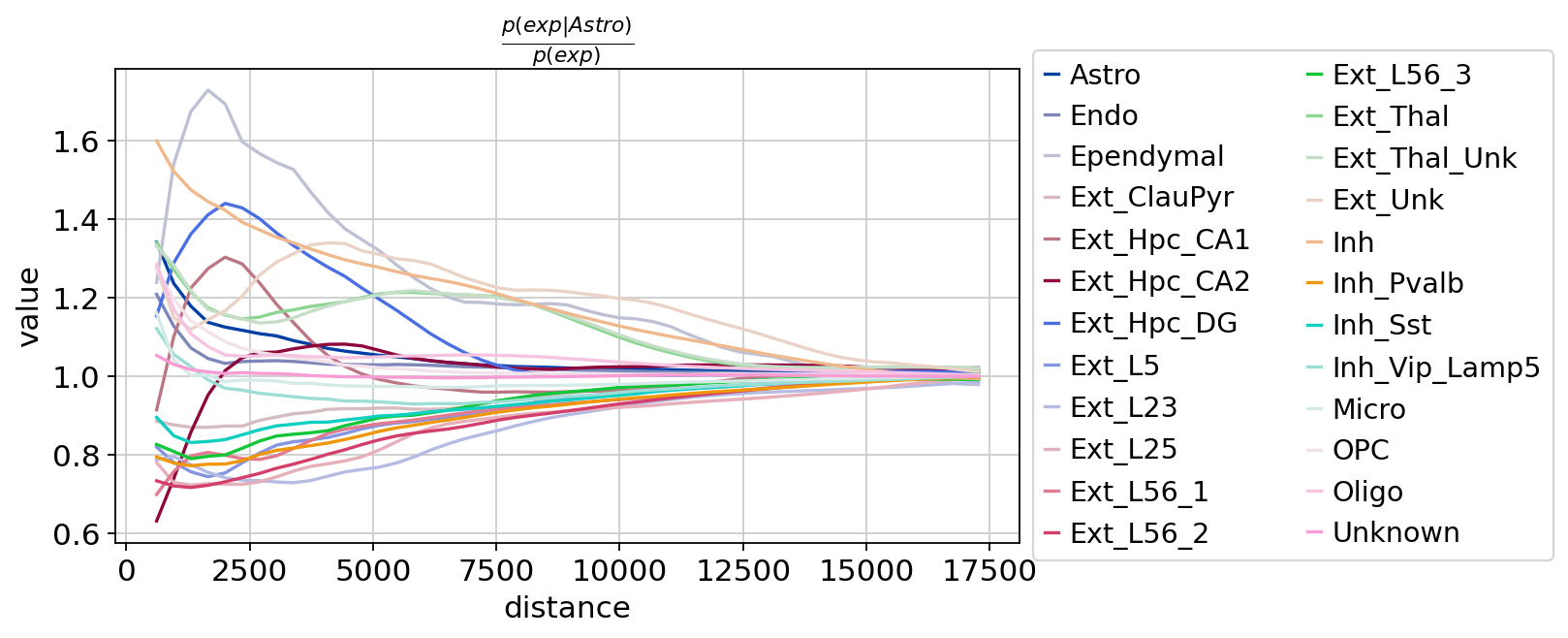
Figure Legend: Cluster co-occurrence score for each cell. In the tissue, as the distance threshold increases, the cluster co-occurrence score for each cell changes gradually. X-axis is distance, Y-axis is co-occurrence score.
Spatially Variable Genes Moran's I
Spatially Variable Genes (SVGs) refer to genes with significant variation in expression patterns across different spatial locations (e.g., different regions in tissue sections, different domains in developmental processes) in spatial transcriptomics data. These genes are usually closely related to tissue microenvironment, cell type distribution, developmental gradients, pathological states, etc., and are key entry points for resolving spatial tissue structure and functional heterogeneity. Next, we can find genes showing spatial patterns. Here, we provide a simple method based on the famous Moran's I statistic.
genes = adata[:, adata.var.highly_variable].var_names.values[:1000]
sq.gr.spatial_autocorr(
adata,
mode="moran",
genes=genes,
n_perms=100,
n_jobs=1,
)adataobs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'Main_CellType', 'Sub_CellType'
var: 'gene_ids', 'feature_types', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne', 'Main_CellType_colors', 'Sub_CellType_colors', 'spatial_neighbors', 'Sub_CellType_nhood_enrichment', 'Sub_CellType_co_occurrence', 'moranI'
obsm: 'X_pca', 'X_umap', 'X_tsne', 'spatial'
varm: 'PCs'
obsp: 'distances', 'connectivities', 'spatial_connectivities', 'spatial_distances'
Variable Gene Assessment Results:
adata.uns["moranI"].head(10)| I | pval_norm | var_norm | pval_z_sim | pval_sim | var_sim | pval_norm_fdr_bh | pval_z_sim_fdr_bh | pval_sim_fdr_bh | |
|---|---|---|---|---|---|---|---|---|---|
| Wfdc2 | 0.469195 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000021 | 0.0 | 0.0 | 0.025987 |
| Dpp10 | 0.339641 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000017 | 0.0 | 0.0 | 0.025987 |
| Tshz2 | 0.315615 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000023 | 0.0 | 0.0 | 0.025987 |
| Hpca | 0.304144 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000017 | 0.0 | 0.0 | 0.025987 |
| Spag17 | 0.298851 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000023 | 0.0 | 0.0 | 0.025987 |
| Fibcd1 | 0.298201 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000024 | 0.0 | 0.0 | 0.025987 |
| Tmem212 | 0.261650 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000011 | 0.0 | 0.0 | 0.025987 |
| Ccdc146 | 0.261100 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000021 | 0.0 | 0.0 | 0.025987 |
| Synpo2 | 0.240745 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000014 | 0.0 | 0.0 | 0.025987 |
| Camk2n1 | 0.233325 | 0.0 | 0.000013 | 0.0 | 0.009901 | 0.000016 | 0.0 | 0.0 | 0.025987 |
Spatial expression of top 5 variable genes on tissue section:
adata.uns["moranI"].index[0:5]#sq.pl.spatial_scatter(adata, color=adata.uns["moranI"].index[0:5])
sc.pl.embedding(adata,"spatial",color=adata.uns["moranI"].index[0:4])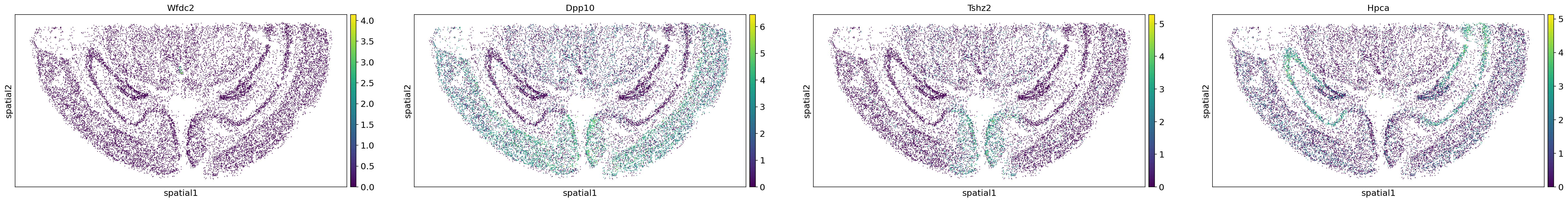
Figure Legend: Expression of four spatially variable genes (Wfdc2, Dpp10, Tshz2, and Hpca) calculated via Moran's I spatial autocorrelation analysis. These genes appear to exhibit distinct pattern characteristics and specific expression for different cell types.
Spatial Dispersion Level of Cell Clusters
Besides neighborhood enrichment scores, we can further investigate spatial organization of cell types in tissue using Ripley's statistics. Ripley's statistics allow analysts to assess whether discrete annotations (e.g., cell types) are aggregated, dispersed, or randomly distributed over the region of interest.
mode = "L"
sq.gr.ripley(adata, cluster_key="Sub_CellType", mode=mode, max_dist=500)adataobs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'Main_CellType', 'Sub_CellType'
var: 'gene_ids', 'feature_types', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'tsne', 'Main_CellType_colors', 'Sub_CellType_colors', 'spatial_neighbors', 'Sub_CellType_nhood_enrichment', 'Sub_CellType_co_occurrence', 'moranI', 'Sub_CellType_ripley_L'
obsm: 'X_pca', 'X_umap', 'X_tsne', 'spatial'
varm: 'PCs'
obsp: 'distances', 'connectivities', 'spatial_connectivities', 'spatial_distances'
adata.uns["Sub_CellType_ripley_L"]0 0.000000 Astro 0.000000
1 10.204082 Astro 1.637580
2 20.408163 Astro 2.315888
3 30.612245 Astro 3.171160
4 40.816327 Astro 3.375957
... ... ... ...n 1195 459.183673 Unknown 60.034669
1196 469.387755 Unknown 61.332776
1197 479.591837 Unknown 62.609327
1198 489.795918 Unknown 63.813106
1199 500.000000 Unknown 64.839684
[1200 rows x 3 columns],
'sims_stat': bins simulations stats
0 0.000000 0 0.000000
1 10.204082 0 0.000000
2 20.408163 0 0.818790
3 30.612245 0 1.157944
4 40.816327 0 1.157944
... ... ... ...n 4995 459.183673 99 20.486117
4996 469.387755 99 20.713929
4997 479.591837 99 21.209662
4998 489.795918 99 21.694070
4999 500.000000 99 22.137633
[5000 rows x 3 columns],
'bins': array([ 0. , 10.20408163, 20.40816327, 30.6122449 ,
40.81632653, 51.02040816, 61.2244898 , 71.42857143,
81.63265306, 91.83673469, 102.04081633, 112.24489796,
122.44897959, 132.65306122, 142.85714286, 153.06122449,
163.26530612, 173.46938776, 183.67346939, 193.87755102,
204.08163265, 214.28571429, 224.48979592, 234.69387755,
244.89795918, 255.10204082, 265.30612245, 275.51020408,
285.71428571, 295.91836735, 306.12244898, 316.32653061,
326.53061224, 336.73469388, 346.93877551, 357.14285714,
367.34693878, 377.55102041, 387.75510204, 397.95918367,
408.16326531, 418.36734694, 428.57142857, 438.7755102 ,
448.97959184, 459.18367347, 469.3877551 , 479.59183673,
489.79591837, 500. ]),
'pvalues': array([[0. , 0.00990099, 0.00990099, ..., 0.00990099, 0.00990099,
0.00990099],
[0. , 0.00990099, 0.00990099, ..., 0.00990099, 0.00990099,
0.00990099],
[0. , 0. , 0.32673267, ..., 0.00990099, 0.00990099,
0.00990099],
...,
[0. , 0.06930693, 0.05940594, ..., 0. , 0. ,
0. ],
[0. , 0.00990099, 0.00990099, ..., 0.00990099, 0.00990099,
0.00990099],
[0. , 0.00990099, 0.00990099, ..., 0.00990099, 0.00990099,
0.00990099]])}
sq.pl.ripley(adata, cluster_key="Sub_CellType", mode=mode)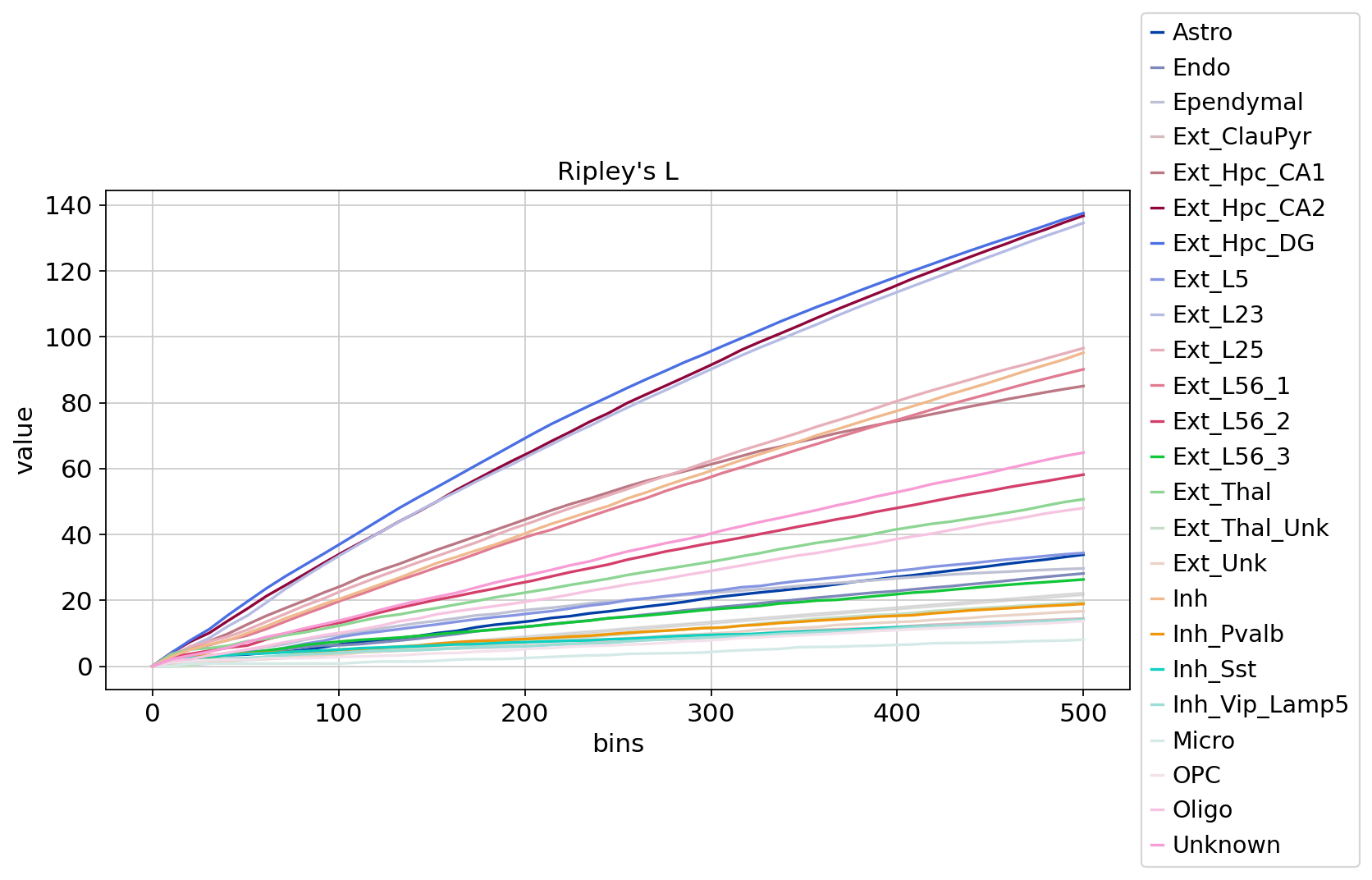
Figure Legend: Change in Ripley's L value as distance increases. X-axis is distance, Y-axis is dispersion level.