stLearn Advanced Spatial Transcriptomics Analysis: Ligand-Receptor Interaction Hotspot Identification
Background Introduction
stLearn uses ligand-receptor co-expression information between neighboring spots and cell type diversity to calculate ligand-receptor (LR) scores. It can identify hotspot regions within a given tissue where LR interactions between cell types are more likely to occur compared to the spatial distribution of random non-interacting gene pairs (i.e., considering communication between neighboring cells and LR co-expression).
stLearn Calculation Results
import stlearn as st
import numpy as np
import pandas as pd
# matplotlib version 3.5.3, higher versions conflict with stlearn
import matplotlib.pyplot as plt
import matplotlib.image as mpimg
import scanpy as sc
import re
import warnings
from PIL import Image
import os@jit(parallel=True, nopython=False)
stLearn Cell Communication Module Input Parameters:
- SAMple_name: Sample name
- meta_path: Metadata file path
- spot_diameter_fullres: Size of cell/spot, related to spatial distance range in analysis steps
- species: Species, must be "human" or "mouse"
- meta_celltype_colums_name: Set column name for cell types in adata object obs
sample_name = "N1"
meta_path = "../../data/AY1748480899609/meta.tsv"
spot_diameter_fullres = 50
species = "human"
meta_celltype_colums_name = "CellAnnotation"- Read SeekSpace data and perform normalization, dimensionality reduction, clustering (input.rds needs to be converted to matrix file beforehand)
adata = sc.read_10x_mtx("./filtered_feature_bc_matrix/")
spatial = pd.read_csv('./filtered_feature_bc_matrix/cell_locations.tsv',sep="\t",index_col=0)
spatial = spatial.loc[:,("x","y")]
selected_rows = spatial.loc[spatial.index.isin(adata.obs_names)]
selected_rows.columns = ["imagecol","imagerow"]
selected_rows = selected_rows.reindex(adata.obs_names)
selected_rows = selected_rows*0.265385
a = st.create_stlearn(count=adata.to_df(),spatial=selected_rows,library_id=sample_name, scale=1,spot_diameter_fullres=spot_diameter_fullres)
a.layers["raw_count"] = a.X
# Preprocessing
st.pp.normalize_total(a)
st.pp.log1p(a)
# Keep raw data
a.raw = a
st.pp.scale(a)
st.em.run_pca(a,n_comps=50,random_state=0)
st.pp.neighbors(a,n_neighbors=25,use_rep='X_pca',random_state=0)
st.tl.clustering.louvain(a,random_state=0)
sc.tl.umap(a)Log transformation step is finished in adata.X
Scale step is finished in adata.X
PCA is done! Generated in adata.obsm['X_pca'], adata.uns['pca'] and adata.varm['PCs']
/jp_envs/envs/stlearn/lib/python3.9/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
2025-08-28 17:39:53.325920: I tensorflow/core/platform/cpu_feature_guard.cc:193] This TensorFlow binary is optimized with oneAPI Deep Neural Network Library (oneDNN) to use the following CPU instructions in performance-critical operations: SSE4.1 SSE4.2 AVX AVX2 AVX512F AVX512_VNNI AVX512_BF16 AVX_VNNI AMX_TILE AMX_INT8 AMX_BF16 FMA
To enable them in other operations, rebuild TensorFlow with the appropriate compiler flags.
2025-08-28 17:39:55.109291: I tensorflow/core/util/port.cc:104] oneDNN custom operations are on. You may see slightly different numerical results due to floating-point round-off errors from different computation orders. To turn them off, set the environment variable \`TF_ENABLE_ONEDNN_OPTS=0\`.
Created k-Nearest-Neighbor graph in adata.uns['neighbors']
Applying Louvain cluster ...n Louvain cluster is done! The labels are stored in adata.obs['louvain']
Add annotated cell types to adata object
cluster_name = "celltype"
celltype = pd.read_csv(meta_path,index_col=0,sep = "\t")
celltype = celltype.loc[a.obs.index]
a.obs[cluster_name] = celltype[meta_celltype_colums_name]
a.obs[cluster_name] = a.obs[cluster_name].astype('category')- Divide spatial slice into length n_, width n_
Note: To save time, divide the spatial slice into n×n grids (equivalent to bins) for subsequent cell communication analysis (grid_step=True); if you want to calculate cell communication at single-cell level, skip this step (grid_step=False)
grid_step=False
if grid_step:
#n_ = 125
print(f'{n_grid} by {n_grid} has this many spots:\n', n_grid*n_grid)
a = st.tl.cci.grid(a,n_row=n_grid, n_col=n_grid, use_label = cluster_name)
print(a.shape)- Load ligand-receptor database, calculate significance of LR expression
lrs = st.tl.cci.load_lrs(['connectomeDB2020_lit'], species=species)
st.tl.cci.run(a, lrs,
min_spots = 20, #Filter out any LR pairs with no scores for less than min_spots
distance=None, # None defaults to spot+immediate neighbours; distance=0 for within-spot mode
n_pairs=10000, # Number of random pairs to generate; low as example, recommend ~10,000
n_cpus=12, # Number of CPUs for parallel. If None, detects & use all available.
)Spot neighbour indices stored in adata.obsm['spot_neighbours'] & adata.obsm['spot_neigh_bcs'].
Altogether 1483 valid L-R pairs
Generating backgrounds & testing each LR pair...: 1%|▏ [ time left: 2:49:41 ]
Ligand-Receptor Expression Significance Results
lr_info = a.uns['lr_summary']lr_info| n_spots | n_spots_sig | n_spots_sig_pval | |
|---|---|---|---|
| THBS1_LRP1 | 3185 | 1505 | 1870 |
| PTPRK_PTPRK | 2622 | 1471 | 2013 |
| A2M_LRP1 | 3078 | 1412 | 1845 |
| VEGFA_GPC1 | 3258 | 1260 | 1897 |
| PTPRM_PTPRM | 2731 | 1203 | 1669 |
| ... | ... | ... | ... |
| FGF3_FGFRL1 | 27 | 12 | 24 |
| GHRH_VIPR1 | 24 | 12 | 24 |
| INHBC_ACVR2A | 25 | 12 | 20 |
| NPPC_NPR3 | 21 | 12 | 20 |
| BMP10_BMPR1A | 21 | 6 | 13 |
1483 rows × 3 columns
- Rows: Ligand-Receptor Pairs
- Column 1 n_spot: Total number of cells/spots with LR interactions without pval filtering
- Column 2 n_spots_sig: Number of cells/spots with significant LR interactions after corrected pval filtering
- Column 3 n_spots_sig_pval: Number of cells/spots with LR interactions after filtering by set pval
st.tl.cci.adj_pvals(a, correct_axis='spot',
pval_adj_cutoff=0.05, adj_method='fdr_bh')Updated adata.obsm[lr_scores]
Updated adata.obsm[lr_sig_scores]
Updated adata.obsm[p_vals]
Updated adata.obsm[p_adjs]
Updated adata.obsm[-log10(p_adjs)]
Ligand-Receptor Diagnostic Plot
A key aspect of LR analysis is controlling for LR expression levels and frequency when identifying significant hotspots. Therefore, diagnostic plots should show little correlation between LR hotspots and expression levels/frequency. The following diagnostics help check this; if not, it suggests a larger number of Permutations may be needed.
st.pl.lr_diagnostics(a, show=False,figsize=(15,5))array([
dtype=object))
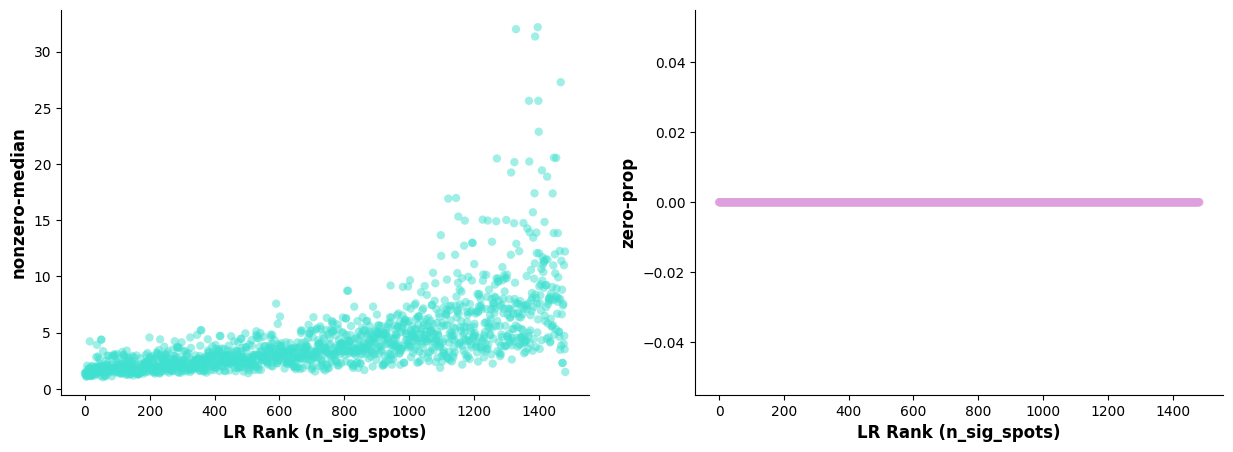
* **Left Plot:** X-axis is sorted LR pairs by n_spots_sig; Y-axis is median expression of LR pairs (non-zero).
* **Right Plot:** X-axis is sorted LR pairs by n_spots_sig; Y-axis is proportion of cells with zero expression of LR pairs.Scatter Plot of Significant Interacting Ligand-Receptors
#st.pl.lr_summary(a, n_top=500,show=False)
st.pl.lr_summary(a, n_top=50, show=False,figsize=(10,3))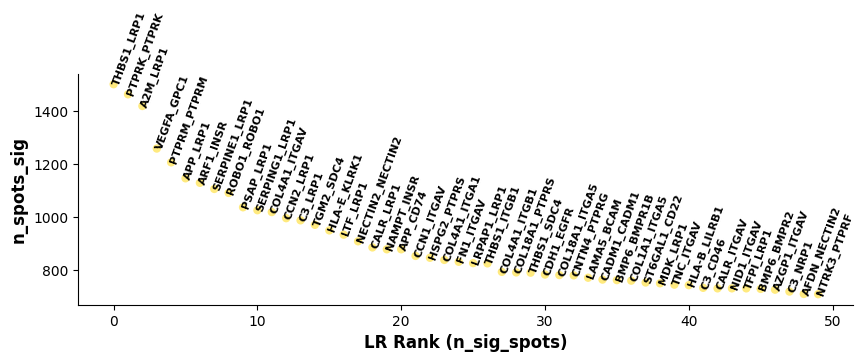
Plot based on lr_summary results: Showing top 50 LR pairs. X-axis represents LR pairs sorted by n_spots_sig; Y-axis represents number of cells/spots with significant LR interaction after corrected pval filtering.Bar Plot of Significant Interacting Ligand-Receptors
st.pl.lr_n_spots(a, n_top=50,max_text=100,show=False,figsize=(11, 3))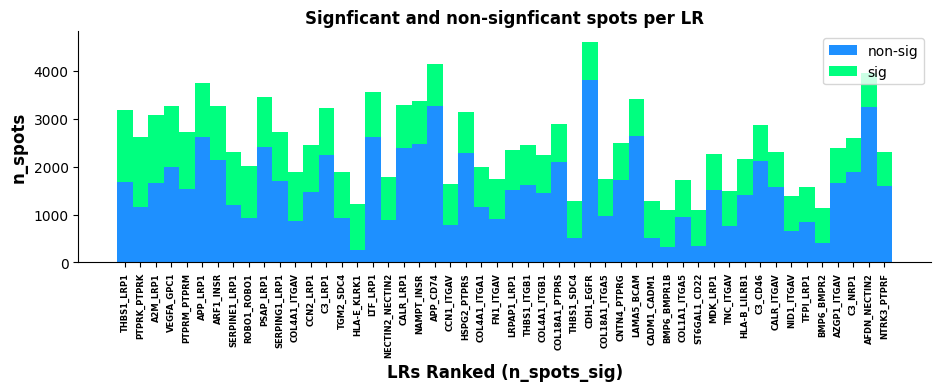
Plot based on lr_summary results: Showing top 500 and top 50 LR pairs. X-axis represents LR pairs sorted by n_spots_sig; Y-axis represents total number of cells/spots with LR interaction (unfiltered by pval); Green and blue represent significant and non-significant LR pairs respectively.LR Statistics Visualization
Visualize hotspots of significant LR pair co-expression to roughly see areas where interactions of interest are concentrated.
lr_scores = pd.DataFrame(a.obsm["lr_scores"])
lr_scores.columns = lr_info.index
lr_scores.index = a.obs.index
p_vals = pd.DataFrame(a.obsm["p_vals"])
p_vals.columns = lr_info.index
p_vals.index = a.obs.index
p_adjs = pd.DataFrame(a.obsm["p_adjs"])
p_adjs.columns = lr_info.index
p_adjs.index = a.obs.index
log10_p_adjs = pd.DataFrame(a.obsm["-log10(p_adjs)"])
log10_p_adjs.columns = lr_info.index
log10_p_adjs.index = a.obs.index
spatial["LR_score"] = list(lr_scores["THBS1_LRP1"])
spatial["p_vals"] = list(p_vals["THBS1_LRP1"])
spatial["p_adjs"] = list(p_adjs["THBS1_LRP1"])
spatial["-log10(p_adjs)"] = list(log10_p_adjs["THBS1_LRP1"])spatial| x | y | LR_score | p_vals | p_adjs | -log10(p_adjs) | |
|---|---|---|---|---|---|---|
| Barcode | ||||||
| AACCTGAGCTAGATTAGAGCGACGGAT_1 | 16898 | 9879 | 0.000000 | 1.0000 | 1.00000 | -0.000000 |
| AAGACAAGCTGATACCTGCTCTGCTTC_1 | 40296 | 7851 | 1.194025 | 0.0111 | 0.01353 | 1.868702 |
| AAGTACCCGAGATAAACAAGGCACTAT_1 | 31685 | 6429 | -0.203235 | 1.0000 | 1.00000 | -0.000000 |
| AGAAGCGGCTTCCGATACCTAATAACG_1 | 16767 | 17485 | -0.460579 | 1.0000 | 1.00000 | -0.000000 |
| AGACTCAATGAATGCACTTCTGCGCTC_1 | 11746 | 14166 | -0.203235 | 1.0000 | 1.00000 | -0.000000 |
| ... | ... | ... | ... | ... | ... | ... |
| TCCGATCCGAAGGAGGGAAGGCACTAT_1 | 30882 | 9777 | -0.314717 | 1.0000 | 1.00000 | -0.000000 |
| TGAGGAGGCTCGACAAGTTCTGCGCTC_1 | 45879 | 10249 | -0.004137 | 1.0000 | 1.00000 | -0.000000 |
| AGGTCTACGATGTGGAGGCTCTGCTTC_1 | 54295 | 6672 | 0.000000 | 1.0000 | 1.00000 | -0.000000 |
| TTCCTTCATGCGCTTCGAAGGCACTAT_1 | 30565 | 10833 | 0.000000 | 1.0000 | 1.00000 | -0.000000 |
| CATAAGCGCTACTCAACGGACTGTGGA_1 | 16847 | 944 | -0.196527 | 1.0000 | 1.00000 | -0.000000 |
20820 rows × 6 columns
# Read spatial HE or DAPI
background_image = mpimg.imread('filtered_feature_bc_matrix/N1_expression.png')
# Create a new figure
fig, ax = plt.subplots(figsize=(12, 8))
# Display background image
ax.imshow(background_image, extent=[0, 55050, 0, 19906], aspect='auto')
# Select numerical column for color gradient (please modify column name based on your data)
# E.g., 'expression_level', 'score', 'intensity', etc.
value_column = 'LR_score' # Please replace with actual numerical column name
# Create colormap
# Choose different colormaps: 'viridis', 'plasma', 'inferno', 'magma', 'coolwarm', 'RdBu_r', etc.
cmap = plt.cm.viridis
norm = plt.Normalize(vmin=spatial[value_column].min(),
vmax=spatial[value_column].max())
# Create scatter plot, color by numerical column
scatter = ax.scatter(spatial['x'], spatial['y'],
c=spatial[value_column],
cmap=cmap, norm=norm,
alpha=0.7, s=15, edgecolors='none')
# Add colorbar
cbar = plt.colorbar(scatter, ax=ax, shrink=0.8)
cbar.set_label(value_column, fontsize=12)
# Add labels and title
ax.set_xlabel('Spatial_1', fontsize=12)
ax.set_ylabel('Spatial_2', fontsize=12)
ax.set_title(f'THBS1_LRP1 {value_column}', fontsize=14)
# Remove axis ticks (optional, for better aesthetics)
ax.set_xticks([])
ax.set_yticks([])
ax.set_aspect("equal")
# Display plot
plt.tight_layout()
plt.show()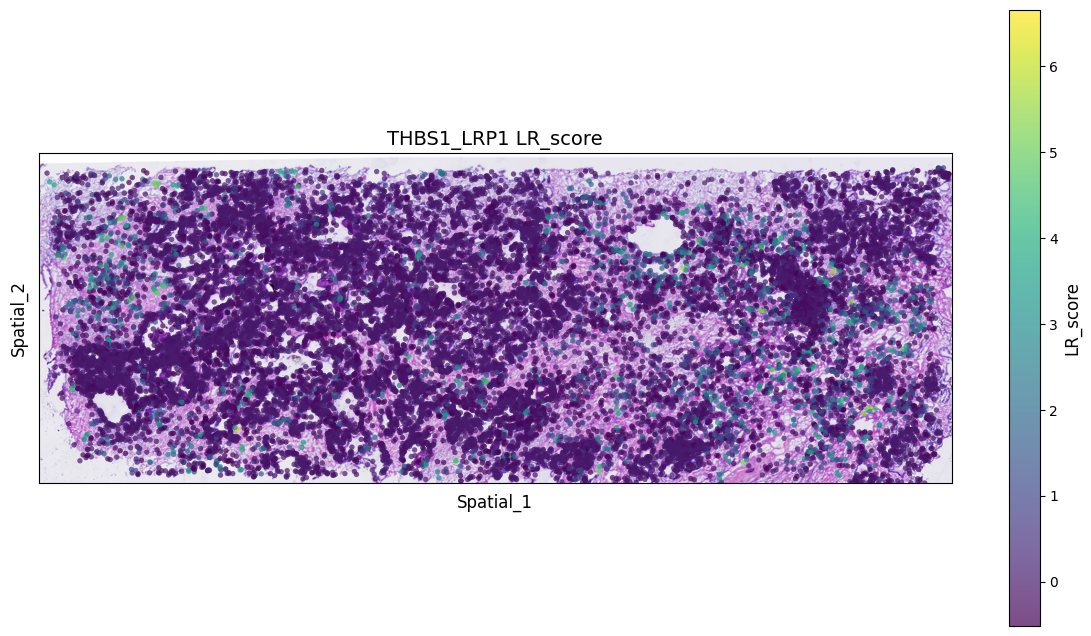
# Read spatial HE or DAPI
background_image = mpimg.imread('filtered_feature_bc_matrix/N1_expression.png')
# Create a new figure
fig, ax = plt.subplots(figsize=(12, 8))
# Display background image
ax.imshow(background_image, extent=[0, 55050, 0, 19906], aspect='auto')
# Select numerical column for color gradient (please modify column name based on your data)
# E.g., 'expression_level', 'score', 'intensity', etc.
value_column = 'p_adjs' # Please replace with actual numerical column name
# Create colormap
# Choose different colormaps: 'viridis', 'plasma', 'inferno', 'magma', 'coolwarm', 'RdBu_r', etc.
cmap = plt.cm.viridis
norm = plt.Normalize(vmin=spatial[value_column].min(),
vmax=spatial[value_column].max())
# Create scatter plot, color by numerical column
scatter = ax.scatter(spatial['x'], spatial['y'],
c=spatial[value_column],
cmap=cmap, norm=norm,
alpha=0.7, s=15, edgecolors='none')
# Add colorbar
cbar = plt.colorbar(scatter, ax=ax, shrink=0.8)
cbar.set_label(value_column, fontsize=12)
# Add labels and title
ax.set_xlabel('Spatial_1', fontsize=12)
ax.set_ylabel('Spatial_2', fontsize=12)
ax.set_title(f'THBS1_LRP1 {value_column}', fontsize=14)
# Remove axis ticks (optional, for better aesthetics)
ax.set_xticks([])
ax.set_yticks([])
ax.set_aspect("equal")
# Display plot
plt.tight_layout()
plt.show()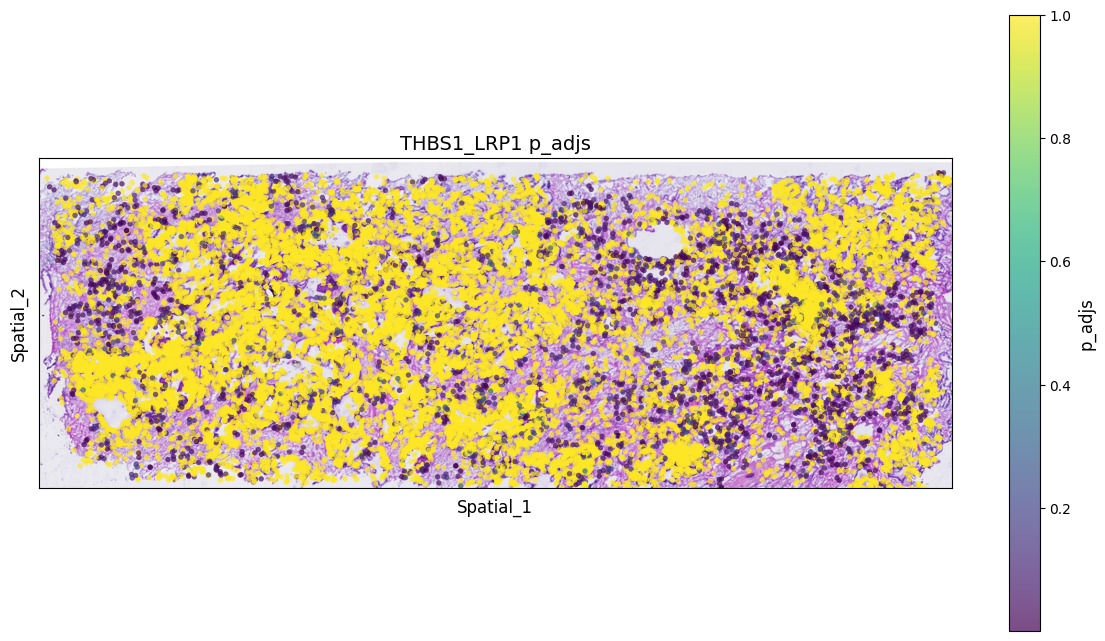
- Predict Significant Interacting Cell Types
st.tl.cci.run_cci(a, cluster_name, # Spot cell information either in data.obs or data.uns
min_spots=3, # Minimum number of spots for LR to be tested.
sig_spots=True, # Only consider neighbourhoods of spots which had significant LR scores.
n_perms=1000, # Permutations of cell information to get background, recommend ~1000
n_cpus=16
)Counting celltype-celltype interactions per LR and permutating 1000 times.: 0%| [ time left: 31:03:47 ]
Cell Type Diagnostic Plot
Check correlation between LR interaction and cell counts of cell types. If permutations are sufficient, charts below should show little or no correlation; otherwise, increase n_perms.
st.pl.cci_check(a, cluster_name,show=False,figsize=(10,4),cell_label_size=10,axis_text_size=12)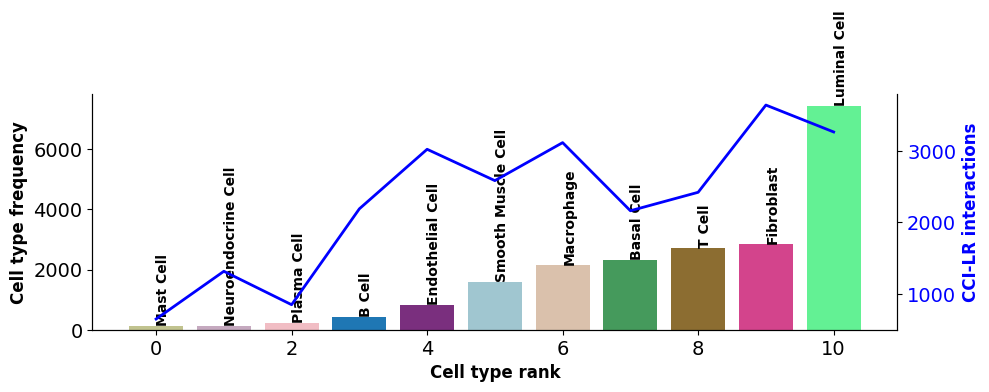
X-axis represents cell types; Bar chart left Y-axis represents cell type count; Line chart right Y-axis represents number of interacting LRs for cell type.CCI Network Plot
st.pl.ccinet_plot(a, cluster_name, return_pos=True,pad=0.3,node_size_exp=0.8)
lr_pair = a.uns['lr_summary'].index.values[0:3]
st.pl.ccinet_plot(a, cluster_name, lr_pair[1], min_counts=2,pad=0.3,node_size_exp=0.8)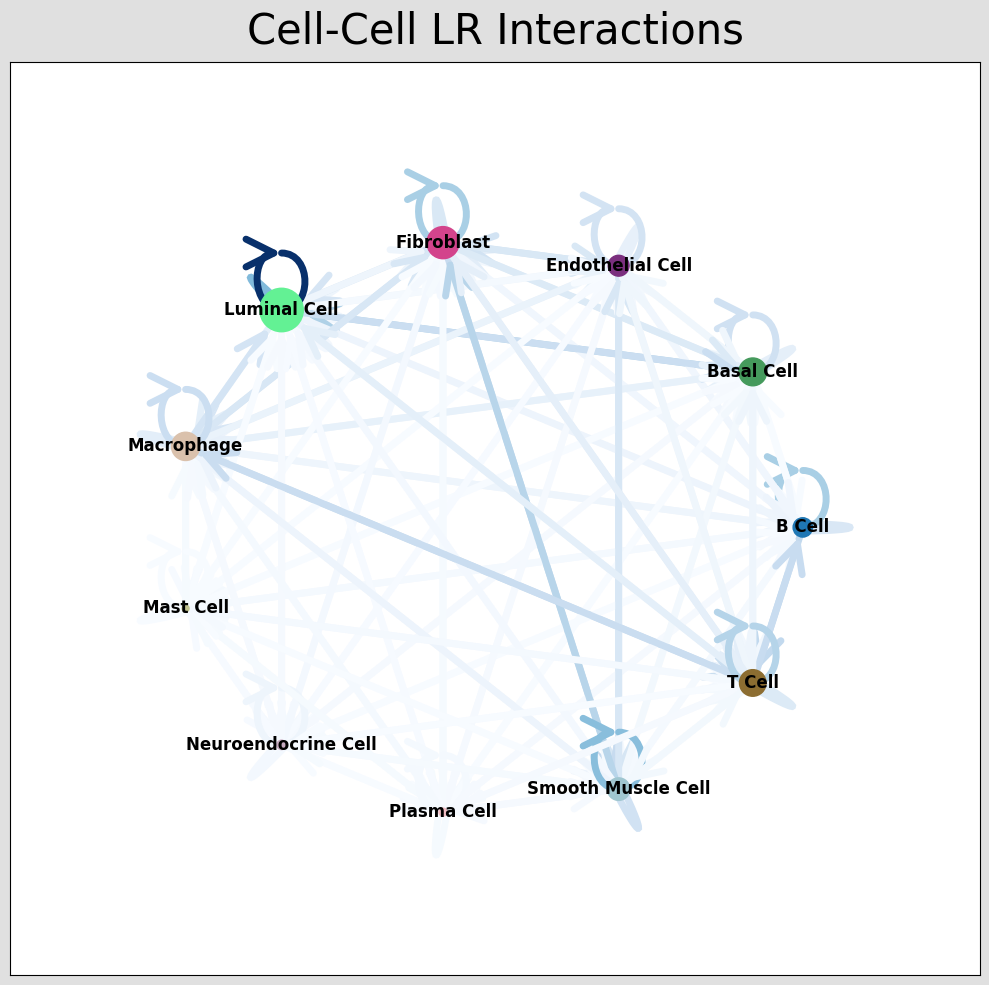
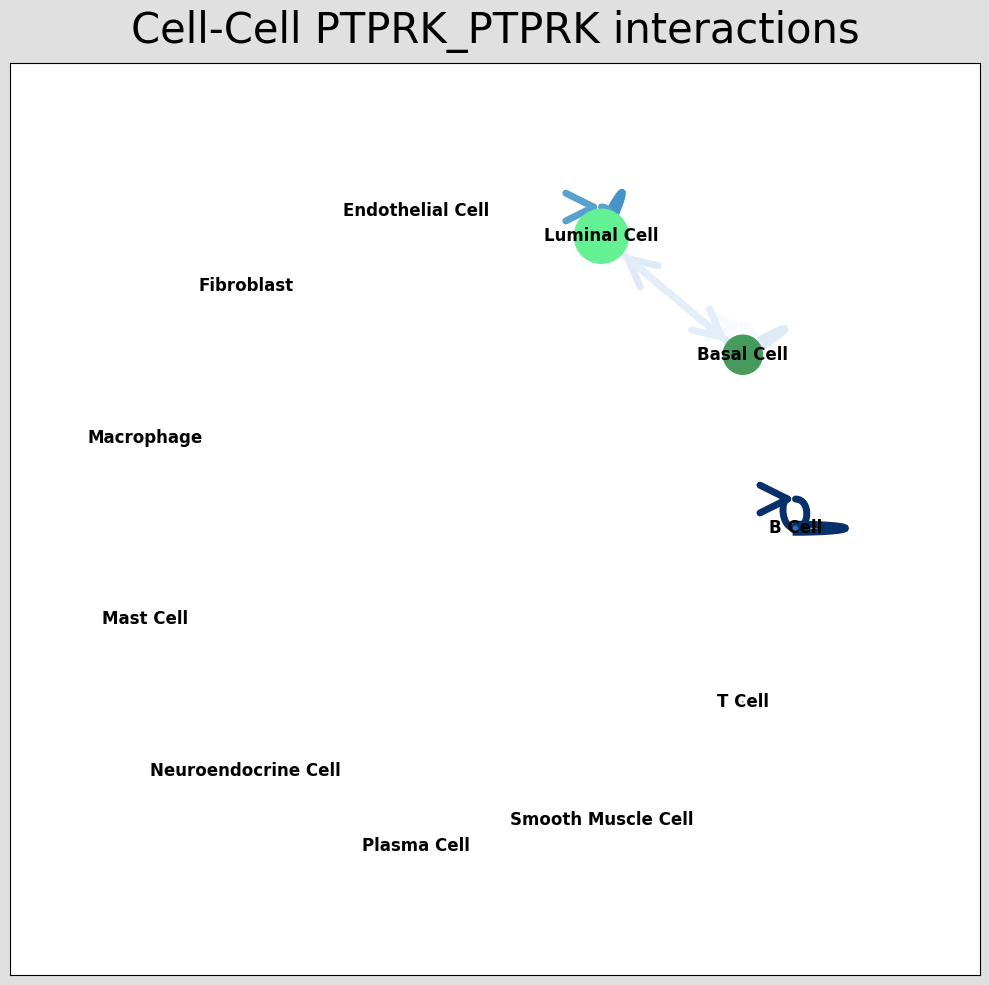
CCI network plot shows cell type interactions for corresponding LR pairs, plotted via per_lr_cci_* results. Each color represents a cell type; lines pointing from a cell type to itself or others represent interactions; line color intensity represents interaction strength.CCI Chord Diagram
st.pl.lr_chord_plot(a, cluster_name,show=False)
st.pl.lr_chord_plot(a, cluster_name,show=False,lr=lr_pair[1])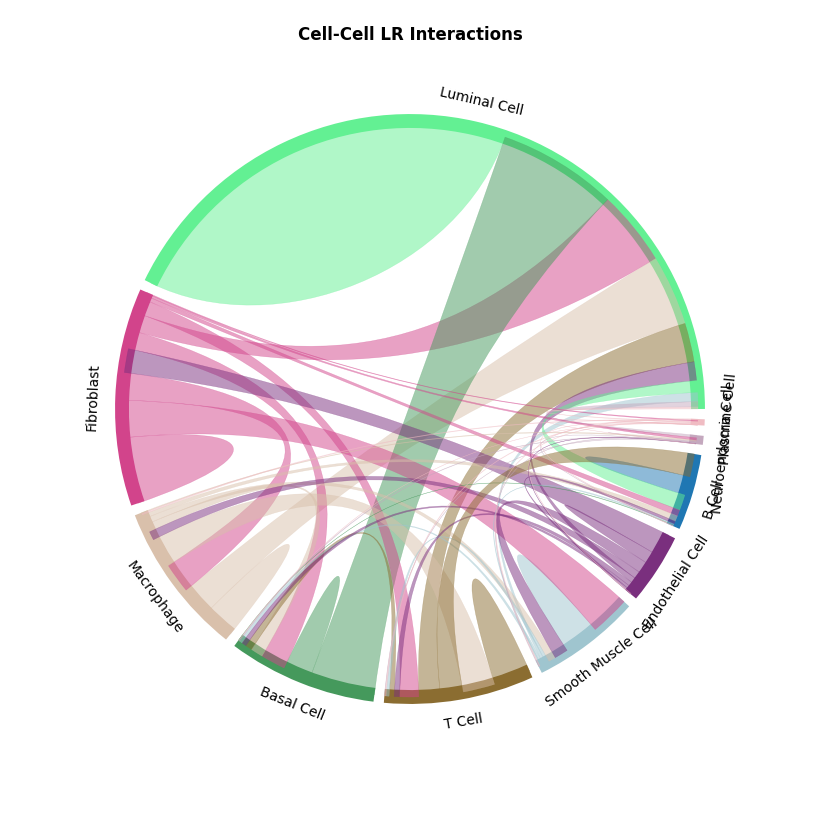
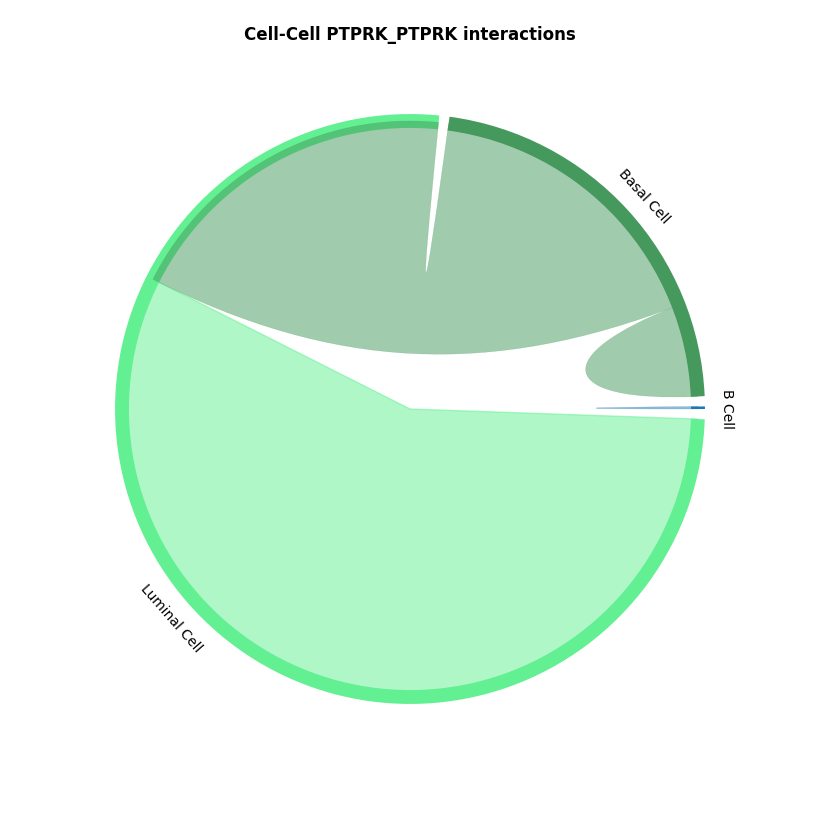
CCI chord diagram shows cell type interactions for corresponding LR pairs, plotted via per_lr_cci_* results. Each color represents a cell type; chord width represents interaction strength, wider chords mean stronger interactions.CCI Heatmap
st.pl.lr_cci_map(a, cluster_name, lrs=None, min_total=100, figsize=(20,4),show=False)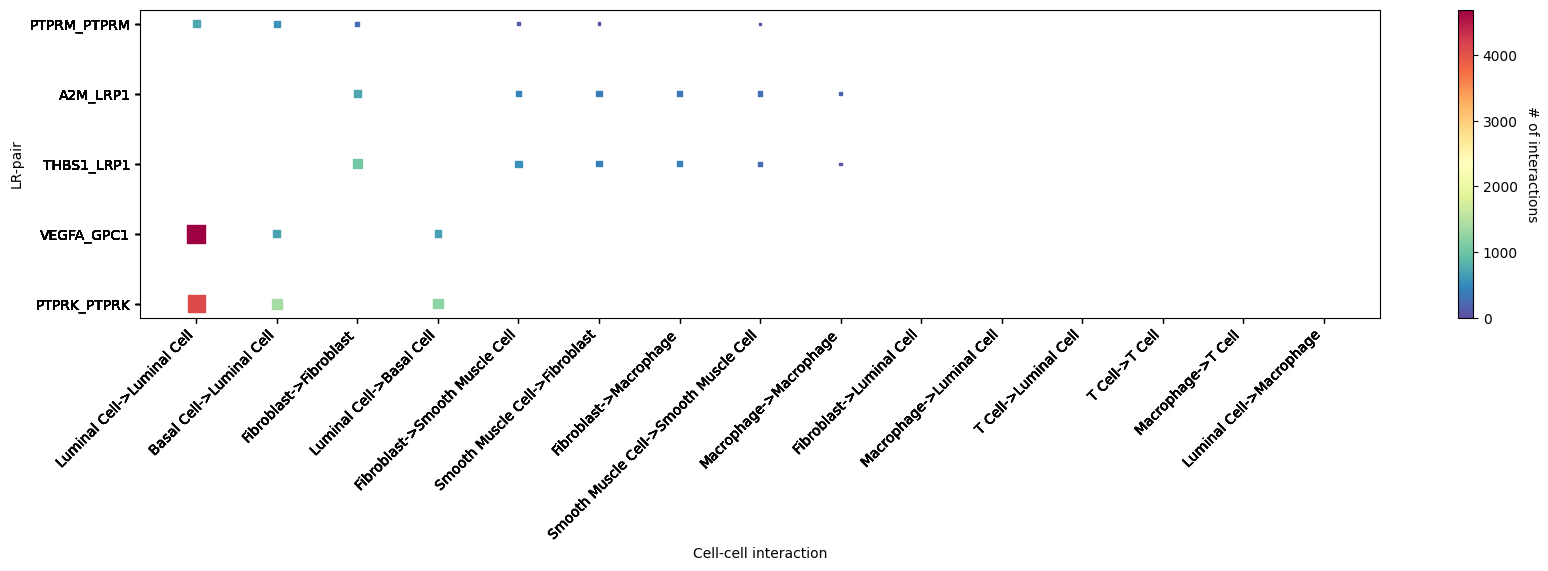
CCI heatmap shows LR interactions corresponding to cell types. X-axis represents interacting cell types; Y-axis represents interacting LRs; color represents interaction strength, redder means stronger. Shows top 5 significantly interacting LR pairs.st.pl.cci_map(a, cluster_name,show=False,figsize=(5,4))
st.pl.cci_map(a, cluster_name, lr=lr_pair[1],show=False,figsize=(5,4))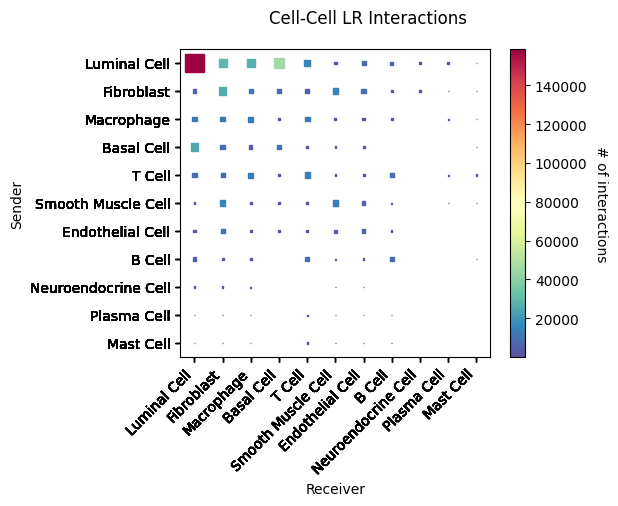
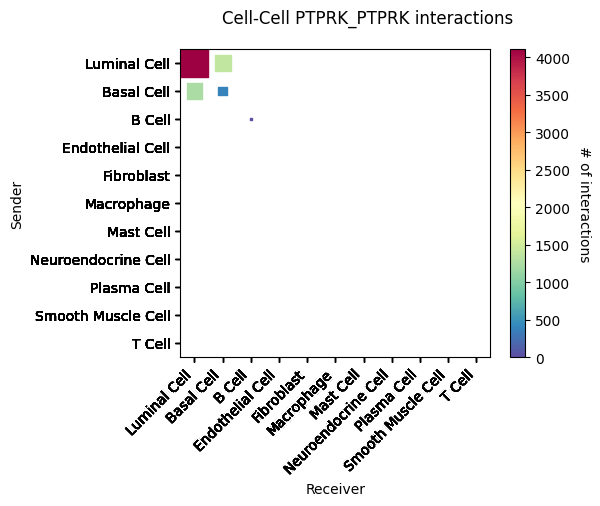
CCI heatmap shows cell type interactions. X and Y axes represent cell types; color represents interaction strength between two cell types.Result Files
lr_diagnostic_plots.png/pdf LR Diagnostic Plot
lr_significant_scatter.png/pdf Significant Interacting LR Scatter Plot
lr_significant_bar.png/pdf Significant Interacting LR Bar Plot
celltype_lr_diagnostic_plots.png/pdf Cell Type LR Diagnostic Plot
CCI_network_plots_*.png/pdf Cell Type Interaction Network Plot
CCI_chord_plots_*.png/pdf Cell Type Interaction Chord Diagram
*CCI_heatmap_plots.png/pdf Cell Type Interaction Heatmap
Literature Case Analysis
- Literature 1:
The article "Spatially organized tumor-stroma boundary determines the efficacy of immunotherapy in colorectal cancer patients" used stlearn to reveal cell type interactions at the tumor-stroma boundary.
References
[1] Pham, D., Tan, X., Balderson, B. et al. Robust mapping of spatiotemporal trajectories and cell–cell interactions in healthy and diseased tissues. Nat Commun 14, 7739 (2023).
[2] Feng, Y., Ma, W., Zang, Y. et al. Spatially organized tumor-stroma boundary determines the efficacy of immunotherapy in colorectal cancer patients. Nat Commun 15, 10259 (2024).
Appendix
.xls, .txt: Result data table files, separated by tabs. Unix/Linux/Mac users use less or more commands to view; Windows users use advanced text editors like Notepad++ or Microsoft Excel.
.png: Result image files, bitmap, lossless compression.
.pdf: Result image files, vector graphics, scalable without distortion, convenient for viewing and editing, use Adobe Illustrator for editing, suitable for publication.