SeekArc Tools v1.0.0
SeekArc Tools v1.0.0 is a software developed by SEEKGENE for processing single-cell ATAC multi-omics data. The software independently processes transcriptome libraries and ATAC libraries for tasks, such as: Barcode extraction and correction, alignment. Quantification of each barcode, joint cell identification, and generation of cell expression matrices for downstream analysis. Enables subsequent clustering and differential analysis, as well as calculation of associations between genes and peaks for downstream interpretation.
Download
SeekArc Tools v1.0.0
Download-SeekArc Tools - md5: af6e054480ca082a554b499fe79c3a48
wget
mkdir seekarctools_v1.0.0
cd seekarctools_v1.0.0
wget -c -O seekarctools_v1.0.0.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seekarctools/seekarctools_v1.0.0.tar.gz"curl
mkdir seekarctools_v1.0.0
cd seekarctools_v1.0.0
curl -C - -o seekarctools_v1.0.0.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seekarctools/seekarctools_v1.0.0.tar.gz"Installation Guide
Installation:
# decompress
tar zxf seekarctools_v1.0.0.tar.gz
# install
source ./bin/activate
./bin/conda-unpack
# export path in bashrc
export PATH=`pwd`/bin:$PATH
echo "export PATH=$(pwd)/bin:\$PATH" >> ~/.bashrcConfirm installation:
seekarctools_py --versionTutorials
Data preparation
Download sample datasets
sample datasets - md5: 45a48572f5ffd00dd6e6d79e3ad495d4(Species:mouse)
wget
wget -c -O demo_data.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/rawdata/demo_data.tar.gz"
# decompress
tar -zxf demo_data.tar.gzcurl
curl -C - -o demo_data.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/rawdata/demo_data.tar.gz"
# decompress
tar -zxf demo_data.tar.gzDownload reference genome
Download-human-reference-GRCh38 - md5: a9ce266ead3d89bc0aa77cfa646ca309
Download-mouse-reference-GRCm38 - md5: c4f2244f1d0560d5d3b4c71da06fd08f
wget
# human
wget -c -O refdata-arc-GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/reference/refdata-arc-GRCh38.tar.gz"
# mouse
wget -c -O refdata-arc-GRCm38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/reference/refdata-arc-GRCm38.tar.gz"
# decompress
tar -zxvf refdata-arc-GRCh38.tar.gzcurl
# human
curl -C - -o refdata-arc-GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/reference/refdata-arc-GRCh38.tar.gz"
# mouse
curl -C - -o refdata-arc-GRCm38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/reference/refdata-arc-GRCm38.tar.gz"
# decompress
tar -zxvf refdata-arc-GRCh38.tar.gzThe assembly of the reference genome refers to How to build reference genome?
Run SeekArc Tools v1.0.0
Run tests
Example 1: Basic usage
Set up the necessary configuration files for the analysis, including sample data paths, reference genome paths, etc. Run the SeekArc Tools v1.0.0 using the following command:
seekarctools_py arc run \
--rnafq1 /path/to/demo/demo_GE_S1_L001_R1_001.fastq.gz \
--rnafq2 /path/to/demo/demo_GE_S1_L001_R2_001.fastq.gz \
--atacfq1 /path/to/demo/demo_arc_S2_L002_R1_001.fastq.gz \
--atacfq2 /path/to/demo/demo_arc_S2_L002_R2_001.fastq.gz \
--samplename demo \
--outdir /path/to/outdir \
--refpath /path/to/reference/GRCh38 \
--include-introns \
--core 16Example 2: Adjusting thresholds for re-running peak calling or cell calling
If the identified peaks or cells under default parameters do not meet requirements, users can adjust parameters to skip preceding steps (e.g., alignment) and re-run the workflow to optimize results while saving time.
seekarctools_py arc retry \
--samplename demo \
--outdir /path/to/outdir \
--refpath /path/to/reference/GRCh38 \
--core 16 \
--qvalue 0.01 \
--snapshift 0 \
--extsize 200 \
--min_len 200 \
--min_atac_count 1000 \
--min_gex_count 500NOTE
Ensure all files from previous runs remain intact and are not deleted or relocated. The --outdir specifies the directory path from the initial SeekArc Tools v1.0.0 analysis. The --min_atac_count must be used together with --min_gex_count; it will not take effect if used alone.
Parameter descriptions
| Parameters | Parameter descriptions |
|---|---|
| --rnafq1 | Paths to R1 fastq files of RNA library |
| --rnafq2 | Paths to R2 fastq files of RNA library |
| --atacfq1 | Paths to R1 fastq files of ATAC library |
| --atacfq2 | Paths to R2 fastq files of ATAC library |
| --samplename | Sample name |
| --outdir | Output directory. Default: ./ |
| --skip_misB | If enabled, no base mismatch is allowed for barcode. Default is 1. |
| --skip_misL | If enabled, no base mismatch is allowed for linker. Default is 1. |
| --skip_multi | If enabled, discard reads that can be corrected to multiple white-listed barcodes. Barcodes are corrected to the barcode with the highest frequency by default. |
| --skip_len | Skip filtering short reads after adapter filter, short reads will be used. |
| --core | Number of threads used for the analysis. |
| --include-introns | When disabled, only exon reads are used for quantification. When enabled, intron reads are also used for quantification. |
| --refpath | The path of reference genome. |
| --star_path | External STAR software path. If the index in the reference genome is built by another STAR, please specify its path. |
| --qvalue | Minimum FDR (q-value) cutoff for peak detection. Default: 0.05. |
| --nolambda | If True, MACS3 will use the background lambda as local lambda. This means MACS3 will not consider the local bias at peak candidate regions. |
| --snapshift | MACS3 peak detection shift size. Default: 0. |
| --extsize | MACS3 peak detection extension size. Default: 400. |
| --min_len | Minimum length of peaks. If not set, it will be set to "extsize". Default: 400. |
| --broad | If enabled, perform broad peak calling and generate results in UCSC gappedPeak format, which encapsulates the nested structure of peaks. |
| --broad_cutoff | Threshold for broad peak calling. Default: 0.1. |
| --min_atac_count | Cell caller override: define the minimum number of ATAC transposition events in peaks (ATAC counts) for a cell barcode. |
| --min_gex_count | Cell caller override: define the minimum number of GEX UMI counts for a cell barcode. |
| -h, --help | Show this parameter descriptions. |
Output descriptions
"Here's the output directory structure: each line represents a file or folder, indicated by "├──", and the numbers indicate three important output files.
./
├── outs
│ ├── filtered_feature_bc_matrix 1
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── filtered_peaks_bc_matrix 2
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── raw_feature_bc_matrix 3
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── raw_peaks_bc_matrix 4
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── demo_A_fragments.tsv.gz 5
│ ├── demo_A_fragments.tsv.gz.tbi 6
│ ├── demo_A_mem_pe_Sort.bam 7
│ ├── demo_A_peaks.bed 8
│ ├── demo_E_SortedByName.bam 9
│ ├── demo.rds 10
│ ├── demo_report.html 11
│ └── demo_summary.csv 12
└── analysis- Transcriptome cell sparse matrix
- ATAC cell sparse matrix
- Transcriptome raw sparse matrix
- ATAC raw sparse matrix
- ATAC per fragment information file
- Fragments file index
- The bam file of ATAC libiary
- ATAC peaks
- The bam file of RNA libiary
- Matrix in rds format
- Final report in html
- Quality control information in csv
Algorithms Overview
RNA library analysis
estep1: barcode/UMI extraction
RNA library structure: 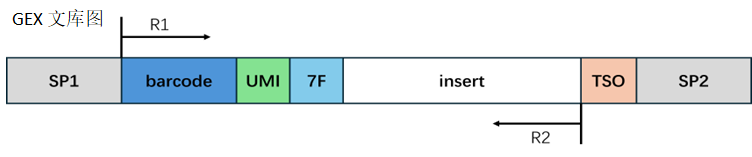
Barcode processing:
The corresponding sequence is extracted based on the structural design. When the extracted barcode sequence is found in the whitelist, it is considered a valid barcode and the read count with valid barcodes is recorded. When the barcode is not found in the whitelist, it is considered an invalid barcode.
TIP
During sequencing process, there is a certain probability that sequencing error occurs. With the presence of a whitelist, SeekArc Tools v1.0.0 are able to do barcode correction. When correction is enabled, if sequences that are one base differ (one humming distance apart) from the invalid barcode appear in the whitelist, we will consider the following circumstances:
- If there is one and only sequence exists in the whitelist, we will correct the invalid barcode to the barcode in the whitelist.
- If multiple sequences exist in the whitelist, we will correct the invalid barcode to the sequence with the highest read support.
Adapter processing:
In the RNA library, the end of Read2 may contain the reverse complement sequence of 7F, which could be an adapter sequence introduced during library preparation. We remove these contaminating sequences, and the length of the trimmed Read2 needs to be longer than the set minimum length to ensure sufficient information for accurate alignment to the genome. If the length of Read2 after trimming is shorter than the minimum length, we consider this read as an invalid read.
estep2: Alignment
Sequence Alignment
- SeekArc Tools v1.0.0 uses an aligner software called STAR to perform splicing-aware alignment of reads to the genome.
- After the alignment procedure has mapped the reads to the genome, SeekArc Tools v1.0.0 uses another software called QualiMap along with a gene annotation transcript file GTF to bucket the reads into exonic, intronic, and intergenic regions.
- Using featureCounts, the reads aligned to the genome were annotated to genes, with different rules such as strand specificity and features used for annotation. When using exon for annotation, a read is annotated to the corresponding gene if over 50% of its bases are aligned to the exon. When using transcripts for annotation, a read is annotated to the corresponding gene if over 50% of its bases are aligned to the transcript.
After the processing procedure described above, the metrics is shown below:
- Reads Mapped to Genome: Fraction of reads that aligned to the genome (including both uniquely mapped reads and multi-mapped reads)
- Reads Mapped Confidently to Genome: Fraction of reads that uniquely mapped to the genome
- Reads Mapped to Intergenic Regions:Fraction of reads that mapped to intergenic regions
- Reads Mapped to Intronic Regions:Fraction of reads that mapped to intronic regions
- Reads Mapped to Exonic Regions:Fraction of reads that mapped to exonic regions
estep3: Counting
UMI counting
SeekArc Tools v1.0.0 extracts group of reads that shared the same barcode from output BAM file and counts the number of UMIs annotated to genes and the number of reads corresponding to each UMI.
- Filter out reads whose UMIs consist of repetitive bases. For example, TTTTTTTT.
- If a read has multiple annotations and only one gene annotation is from exon, it is considered a valid read, and all others are filtered.
UMI correction:
NOTE
UMIs may also have sequencing errors during sequencing process. By default, SeekArc Tools v1.0.0 uses the adjacency from UMI-tools to correct UMIs.
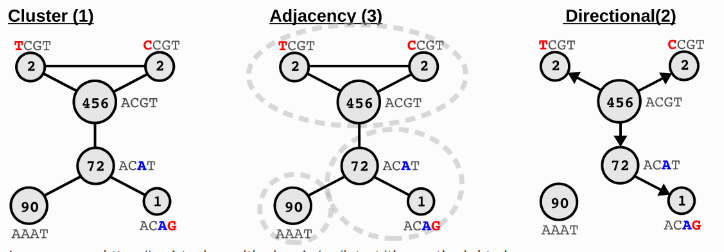
Image source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
ATAC library analysis
astep1: Barcode extraction
Atac library structure: 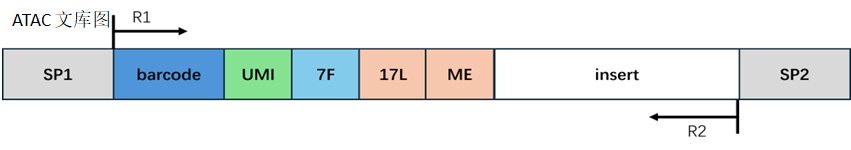
Barcode processing:
Same processing method as estep1.
Adapter and reads processing:
In the Atac library, Read1 may contain a fixed sequence at the end, which could include adapter sequences; the end of Read2 may contain the reverse complement of the fixed sequence and adapter sequences. We remove these contaminating sequences, and the length of the trimmed Read1 or Read2 needs to be longer than the set minimum length to ensure sufficient information for accurate alignment to the genome. If the length of Read1 or Read2 after trimming is shorter than the minimum length, we consider this read as an invalid read; if the length of Read1 or Read2 after trimming is greater than 50, we truncate the first 50bp of the Read1 or Read2 sequence; if the length of Read1 or Read2 after trimming is less than 50 but greater than the minimum length, we retain the entire sequence of Read1 or Read2.
estep2: Alignment
Sequence Alignment:
Use bwa mem to align the processed Read1 and Read2 to the reference genome.
astep3: Counting
Use SnapATAC2 v2.8.0 to process the BAM file output from astep2.
- Read the BAM file to obtain ATAC sequencing and mapping metrics.
- Generate the raw fragments file from the BAM file. Filter out mitochondria fragments to obtain the filtered fragments file.
- Generate an Anndata object from the filtered fragments file.
- Plot the distribution of TSS score.
- Use MACS3 to call peaks, filter duplicate peaks, and output to peaks.bed.
- Generate raw_peaks_bc_matrix dir based on peaks.
- Calculate metrics for each barcode and output to per_barcode_metrics.csv.
- Plot the distribution of insert sizes.
- Cell calling
- Generate filtered_feature_bc_matrix and filtered_peaks_bc_matrix.
- Calculate ATAC library metrics for cells and targeting.
Cell calling
IMPORTANT
If --min_atac_count and --min_gex_count are specified:
- Then determine the barcode as a cell if the UMI count is greater than the min_gex_count value and the events count is greater than the min_atac_count value.
If not specified:
- First, retain barcodes where the UMI count is greater than 1 or the events in peaks count is greater than 1.
- Then, filter barcodes where "the fraction of fragments overlapping called peaks" is less than "the fraction of genome in peaks".
- Deduplicates. Filter barcodes with identical UMI values and events in peaks values.
- Perform log10 transformation on the UMI values and events in peaks values of the filtered barcodes.
- Based on the transformed UMI values and events in peaks values, use the KMeans algorithm to divide the barcodes into two clusters. The cluster with the higher mean is identified as cells, while the other is not.
- Map the barcodes filtered out during deduplication to cells or non-cells based on the determination of the remaining barcodes.
astep4: Subsequent analysis
After the above steps, with the obtained cell expression matrix and ATAC matrix, we can proceed to the next stage of analysis.
Use Signac v1.14.0 to merge the cell expression matrix and ATAC matrix into a single Seurat object, Filter out features not in the database, then normalize the expression matrix and ATAC matrix separately, identify highly variable genes, perform dimensionality reduction and clustering, find differential genes, conduct WNN clustering, and finally calculate feature links.
Release notes
v1.0.0
Initial release
- Release of a stable version supporting standardized analysis workflow for ATAC multi-omics data.