混合物种 scRNA-seq 分析指南:原理、结果解读与常见问题
NOTE
重要前提说明
- SeekSoul Tools 软件没有针对混合物种的特有算法,混合物种样本建议转换为 10x 白名单格式,使用 Cell Ranger 进行分析
- 多组学 ATAC 不支持混合物种分析:10x 软件
cellranger arc和cellranger ATAC均不支持混合物种,本指南仅适用于普通 RNA 文库 - Barcode 转换工具:参考钉盘上生信对外资料中的
Barcode-Converter小工具
构建混合物种参考基因组
数据准备
NOTE
文件格式要求
- 推荐使用 Ensembl 数据库 提供的 GTF 和 FASTA 文件(包含可选标签,便于后续过滤)
- 如果 Ensembl 中不可用,也可使用其他来源的 GTF 和 FASTA 文件
- 必须使用 GTF 格式,不支持 GFF 格式
- GTF/GFF 格式说明参考:Ensembl GFF/GTF 格式文档
过滤非编码基因(推荐)
GTF 文件中可能包含 non-polyA transcripts 相关的条目,这些转录本与蛋白质编码基因存在 overlap。由于这些 overlap 的注释信息,reads 可能会被标记为映射到了多个基因(multi-mapped),从而不会用于后续基因定量。
建议使用 cellranger mkgtf 命令过滤非编码基因:
cellranger mkgtf input.gtf output.gtf --attribute=gene_biotype:protein_coding构建混合物种索引
使用 cellranger mkref 命令构建混合物种参考基因组,需要为每个物种指定 --genome、--fasta 和 --genes 参数:
cellranger mkref \
--genome=GRCh38 \
--fasta=/human/refdata-gex-GRCh38-2020-A/fasta/genome.fa \
--genes=/human/refdata-gex-GRCh38-2020-A/genes/genes.gtf \
--genome=Macaca_fascicularis \
--fasta=/Macaca_fascicularis_GCF_037993035.1/fasta/genome.fa \
--genes=/Macaca_fascicularis_GCF_037993035.1/genes/genes.gtf \
--maxjobs 50 \
--localmem 60 \
--localcores 12验证索引构建
构建完成后,检查 reference.json 文件,确保包含 2 个或以上 的基因组条目,Cell Ranger 才能识别为混合物种:
{
"fasta_hash": "6a2ba19a695f279ec863f521fb1d834d01905c19",
"genomes": [
"GRCh38",
"Macaca_fascicularis"
],
"gtf_hash.gz": "24cb2b8556e5c3740b0816f965c7912f679b17f5",
"input_fasta_files": [
"genome.fa",
"genome.fa"
],
"input_gtf_files": [
"genes.gtf",
"genes.gtf"
],
"mem_gb": 16,
"mkref_version": "8.0.0",
"threads": 2,
"version": null
}NOTE
外源插入基因处理
如果有外源插入基因,可以先将这些基因添加到任意一个物种的基因组和注释文件中,然后再构建混合物种索引。
分析流程与原理
核心原理概述
Cell Ranger 根据每个条形码 (barcode) 比对到不同物种的 UMI 计数进行物种分类:
- 初步分类:根据哪个物种的 UMI 计数更多,将条形码初步分类为对应物种
- Multiplet 识别:如果某个条形码在两个物种的 UMI 计数都超过了各自分布的第 10 百分位数,则判定为多细胞混合体 (multiplet)
NOTE
算法适用性
该识别算法在混合物种细胞比例接近 1:1(如 50:50)时准确性较高,偏离越大,准确性会降低。
详细算法步骤
步骤 1:收集并分析 UMI 计数
对于每一个细胞条形码(barcode),Cell Ranger 统计:
- 物种 A 的 UMI 计数:比对到物种 A 参考基因组的 UMI 数量
- 物种 B 的 UMI 计数:比对到物种 B 参考基因组的 UMI 数量
基于此,每个条形码可归为以下三类之一:
- 物种 A 主导(A > B):比对到物种 A 的 UMI 数量 > 比对到物种 B 的数量
- 物种 B 主导(B > A):比对到物种 B 的 UMI 数量 > 比对到物种 A 的数量
- 两者相当或混合(A ≈ B):可能为双物种混合体
步骤 2:设定分类阈值(Threshold)
为了区分"真正的单细胞"与"可能的多细胞",Cell Ranger 采用**第 10 百分位数(10th percentile)**作为分类临界值:
设定物种 A 细胞的阈值:
- 从所有"物种 A UMI > 物种 B UMI"的条形码中
- 找出这些条形码的物种 A UMI 计数的第 10 百分位数,记为
A_threshold - 任何 barcode,如果其物种 A UMI 数 >
A_threshold,则初步认为是物种 A 单细胞(A singlet)
设定物种 B 细胞的阈值:
- 从所有"物种 B UMI > 物种 A UMI"的条形码中
- 找出这些条形码的物种 B UMI 计数的第 10 百分位数,记为
B_threshold - 任何 barcode,如果其物种 B UMI 数 >
B_threshold,则初步认为是物种 B 单细胞(B singlet)
步骤 3:识别多细胞混合体(Multiplet)
如果某个 barcode 同时满足以下条件,则判定为 multiplet:
- 物种 A UMI 数 >
A_threshold且 - 物种 B UMI 数 >
B_threshold
算法流程总结
对于每个条形码:
统计 speciesA_UMI_count 和 speciesB_UMI_count
如果 speciesA_UMI > speciesB_UMI:
归为"物种 A 主导"
→ 用所有"物种 A 主导"条形码,取 speciesA_UMI 的第 10 百分位 → A_threshold
如果 speciesB_UMI > speciesA_UMI:
归为"物种 B 主导"
→ 用所有"物种 B 主导"条形码,取 speciesB_UMI 的第 10 百分位 → B_threshold
判断逻辑:
如果 speciesA_UMI > A_threshold 且 speciesB_UMI > B_threshold:
→ 判定为 multiplet (双物种混合体)
如果只有 speciesA_UMI 高 → 可能为物种 A singlet
如果只有 speciesB_UMI 高 → 可能是物种 B singlet
(注意:无法区分同物种的双细胞)结果解读
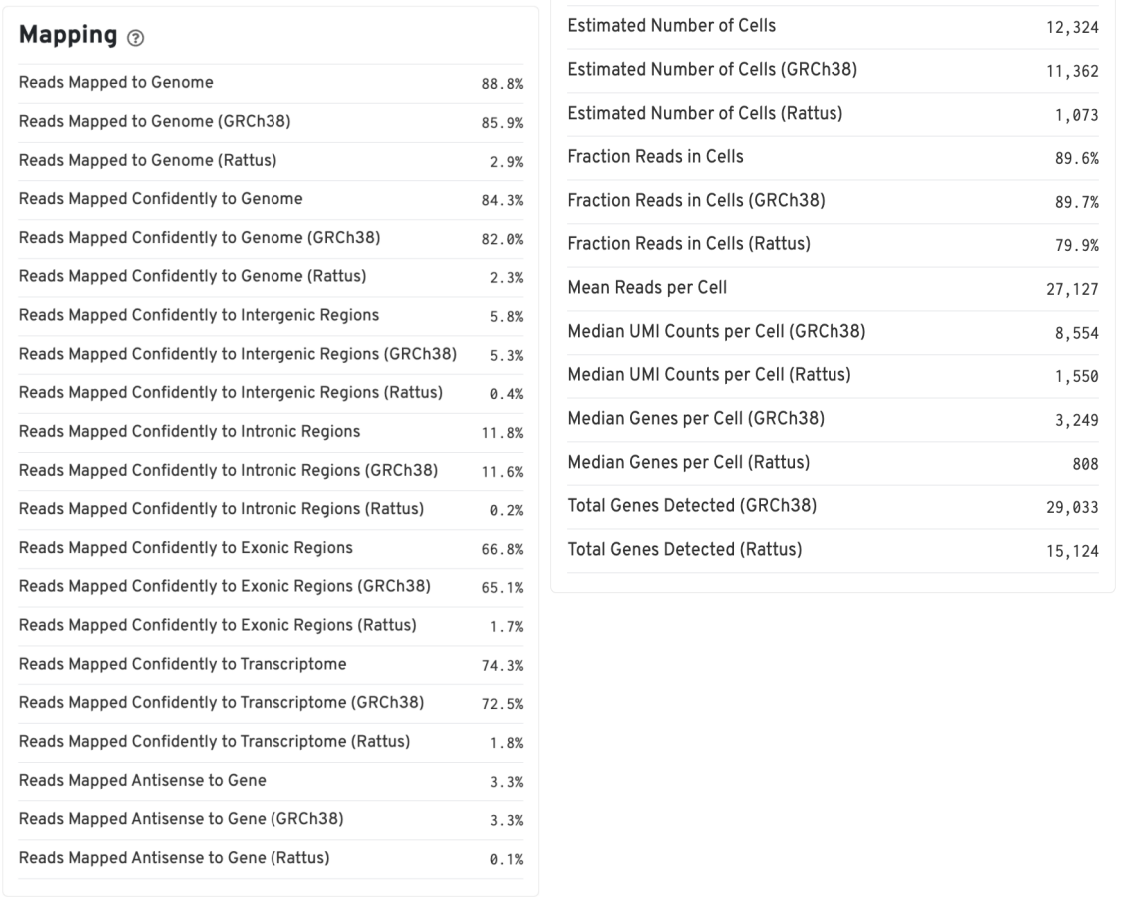
Summary 报告
Summary 报告提供了以下关键信息:
- 各物种的比对率:反映数据质量
- 软件判定的细胞数目:各物种检测到的细胞数量
- 基因中位数:每个细胞的基因表达中位数
Gene Expression 报告
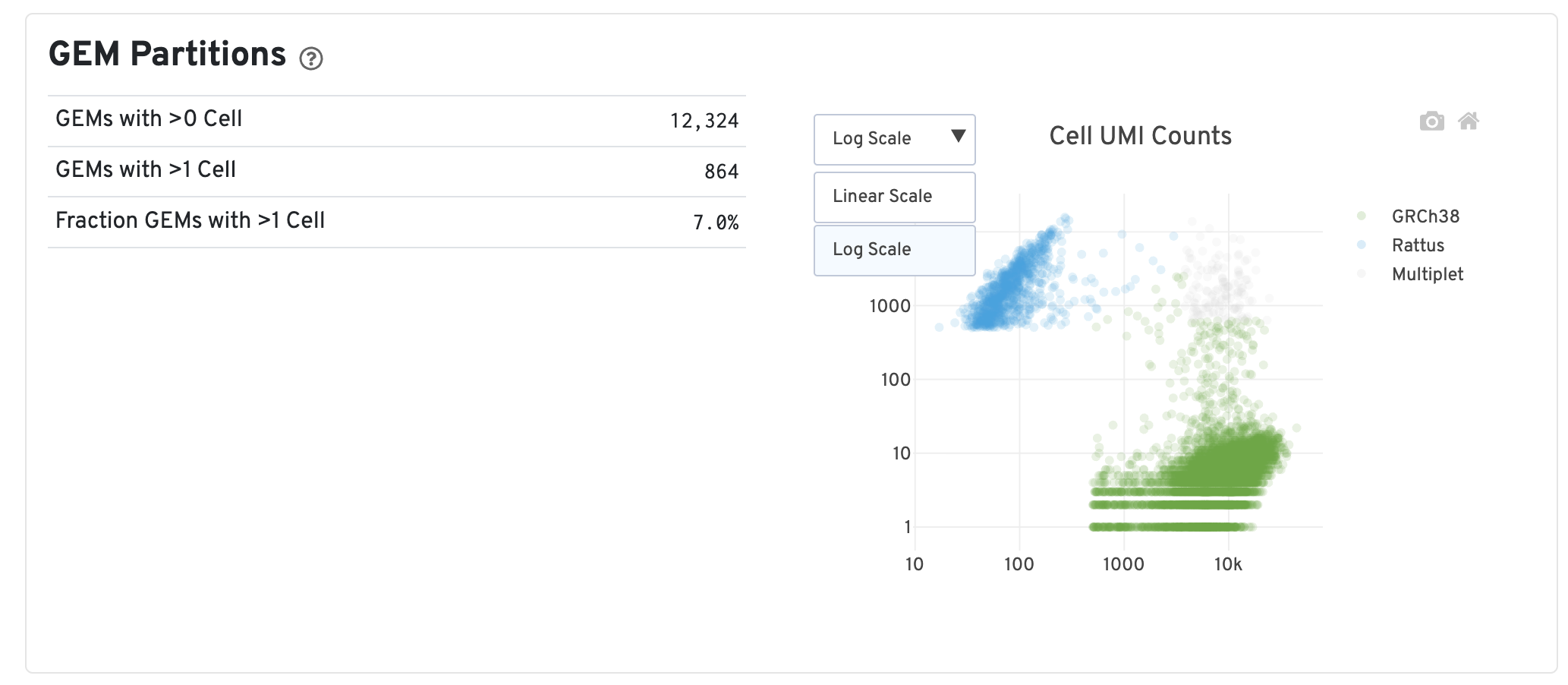
关键指标说明
| 指标 | 定义 | 解读 |
|---|---|---|
| GEMs with >0 Cell | 经过判断,至少有一个细胞的 barcode 数目 | 表示检测到的有效细胞数量 |
| GEMs with >1 Cell | 经过判断,至少有 2 个细胞的 barcode 数目 | 表示多重条形码(multiplets)的数量,比例高表明某些条形码中可能存在多个细胞的 RNA |
| Fraction GEMs with >1 Cell | 估计与多个细胞相关联的 barcode 的平均比例 | 通过自助抽样(bootstrap sampling)计算,并调整了两种细胞类型的比例。反映在所有 barcode 中,有多少比例的 barcode 被推断为与多个细胞相关联 |
| Cell UMI Counts Plot | 每个点代表一个 barcode,坐标轴测量映射到每个物种的总 UMI 计数 | 点的颜色表示与每个 barcode 相关联的推断细胞数量,可直观看到哪些 barcode 可能是 multiplets |
输出文件
常规输出文件
与常规 RNA 分析结果一致,包括:
filtered_feature_bc_matrix/:过滤后的表达矩阵raw_feature_bc_matrix/:原始表达矩阵analysis/:分析结果目录metrics_summary.csv:质控指标汇总
特殊文件:gem_classification.csv
该文件记录了每个 barcode 检测到的各个物种的 UMI 数目和软件最终判断的物种分类:
- 列说明:
barcode:细胞条形码[物种 A 名称]:比对到物种 A 的 UMI 计数[物种 B 名称]:比对到物种 B 的 UMI 计数call:软件判定的物种分类(物种 A 名称、物种 B 名称或 Multiplet)
示例:
barcode,GRCh38,Rattus,call
AAACCCAAGAAACACT-1,44666,22,GRCh38
AAACCCAAGAAACCAT-1,5856,8,GRCh38
AAACCCAAGAAACCCA-1,35589,10,GRCh38
AAACCCAAGAAACCCG-1,37726,13,GRCh38
AAACCCAAGCGATCTC-1,296,14235,Rattus
AAACCCAAGCGCACTT-1,271,15742,Rattus
AAACCCAAGGCGGACT-1,276,12822,Rattus
AAACCCAAGGCTCACC-1,246,13719,Rattus
AAACCCACAATCCCTC-1,2978,8727,Rattus
AAACGAAAGTGAGTGC-1,4600,839,Multiplet
AAACGAAAGTGATGGC-1,3827,983,Multiplet
AAACGAAAGTGCGATA-1,5605,907,Multiplet
AAACGAACAAACCCGC-1,4674,940,Multiplet下游分析:物种细胞判断
有两种分析思路可用于物种细胞判断和拆分:
方法一:使用 Cell Ranger 判定结果
直接使用 gem_classification.csv 中 Cell Ranger 软件判定的物种分类:
# 读取 Cell Ranger 判定结果
cellranger <- read_csv("gem_classification.csv") %>%
select(barcode, call) %>%
rename("cellranger" = "call")
cellranger$barcode <- as.character(cellranger$barcode)
# 合并到 Seurat 对象
ob@meta.data <- ob@meta.data %>%
tibble::rownames_to_column(var = "barcode") %>%
left_join(cellranger, by = "barcode") %>%
tibble::column_to_rownames(var = "barcode")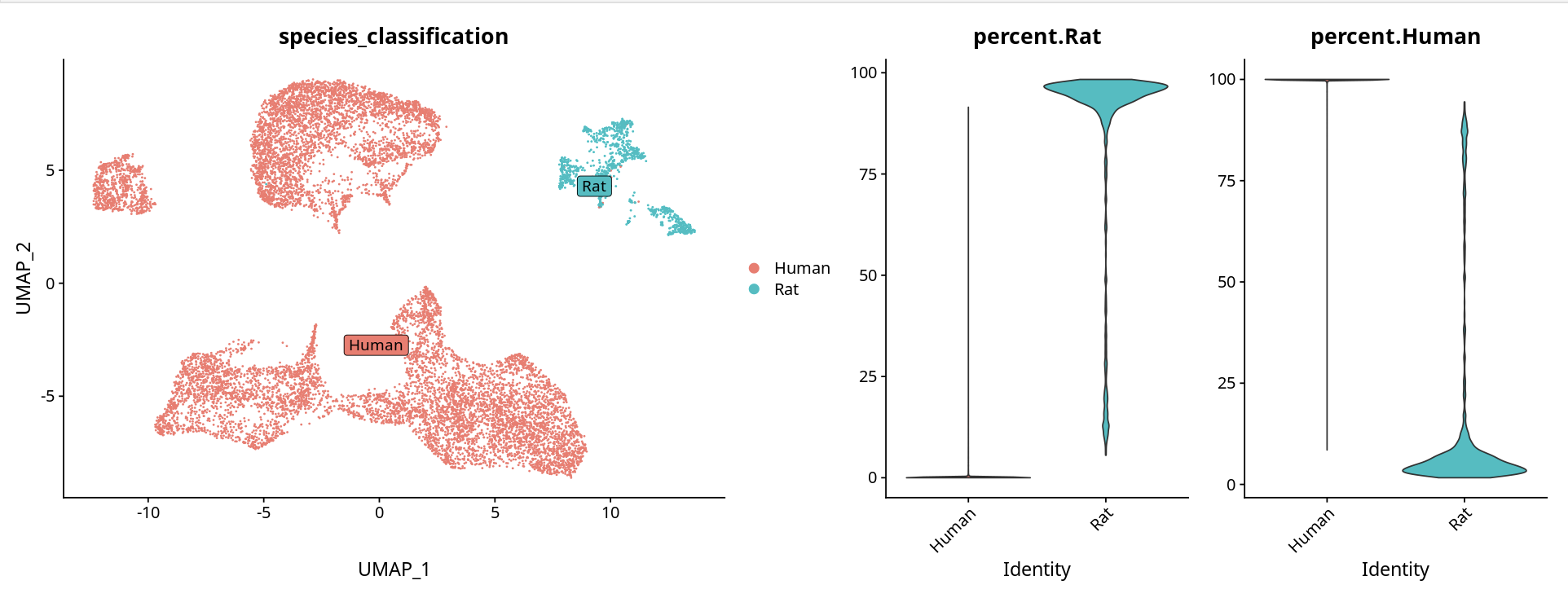
方法二:根据基因表达百分比判断
根据每个cluster表达对应物种基因的百分比进行判断:
# 计算各物种基因表达百分比
ob[['percent.Rat']] <- PercentageFeatureSet(object = ob, pattern = '^Rattus')
ob[['percent.Human']] <- PercentageFeatureSet(object = ob, pattern = '^GRCh38')
# 可视化
p <- DimPlot(ob, label = T, repel = T, label.box = T)
p1 <- VlnPlot(ob, features = c("percent.Rat", "percent.Human"), pt.size = 0)
options(repr.plot.height = 6, repr.plot.width = 16)
p + p1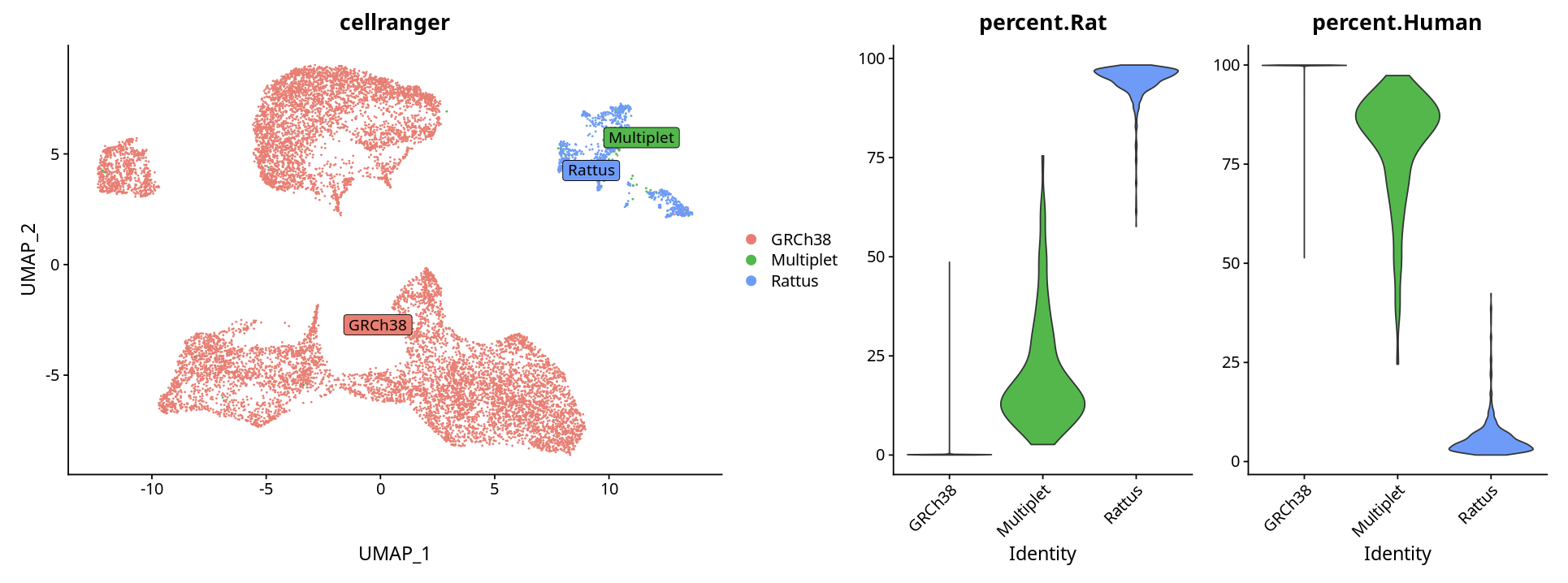
NOTE
云平台工具支持
云平台上"我的工具"中有 SpeciesSplit 模块,可以辅助进行物种拆分分析。
常见问题与故障排除
Q1:如何判断分析结果中是否包含对应物种的细胞
判断方法:
查看质控报告中的比对率: 301→ - 检查各物种的比对率 (Mapping Rate)
- 如果某个物种的比对率非常低(如 < 5%),说明该物种的细胞可能未被捕获
检查UMI和基因计数:
- 如果物种比对率低,检测到的UMI和基因数量一定非常少
- 最终判定的该物种细胞数量不可靠,说明样本中可能没有捕获到对应物种的细胞
查看
gem_classification.csv:- 检查各物种的 UMI 计数分布
- 如果某个物种的 UMI 计数普遍极低,说明该物种细胞可能不存在
故障排除步骤:
# 读取分类结果并检查
classification <- read_csv("gem_classification.csv")
# 统计各物种的细胞数量
table(classification$call)
# 检查各物种的 UMI 计数分布
summary(classification$GRCh38) # 替换为实际物种名称
summary(classification$Rattus) # 替换为实际物种名称Q2:如何理解样本中的 Multiplet 判定
理解要点:
算法局限性:
- Cell Ranger 提供的是固定算法,基于第 10 百分位数阈值
- 在细胞比例接近 1:1 时准确性较高,偏离越大准确性越低
综合判断方法:
- 结合样本的整体聚类结果
- 检查这些细胞的基因表达情况
- 计算对应物种基因的表达百分比
- 检查这些细胞的 gene 和 UMI 的整体情况
- 可结合下游的双细胞预测软件 (如
DoubletFinder、scDblFinder等) 进行验证
验证代码示例:
# 提取multiplet细胞
multiplet_cells <- rownames(ob@meta.data)[ob@meta.data$cellranger == "Multiplet"]
# 检查multiplet细胞的基因表达特征
multiplet_data <- ob@meta.data[multiplet_cells, ]
summary(multiplet_data$nFeature_RNA)
summary(multiplet_data$nCount_RNA)
# 检查物种基因表达百分比
if ("percent.Human" %in% colnames(ob@meta.data)) {
summary(multiplet_data$percent.Human)
summary(multiplet_data$percent.Rat)
}Q3:使用混合物种参考基因组分析的样本数据可以与单物种参考基因组分析的样本进行整合分析吗
答案:可以,但需要预处理。
原因:
使用混合物种参考基因组时,两个物种的基因组都做了标记。例如,基因名称中都加了物种前缀:
- 人类基因:
GRCh38_CD79A - 小鼠基因:
mm10_Actb
整合步骤:
拆分表达矩阵:
- 根据物种前缀拆分表达矩阵
- 将混合物种数据拆分成两个单物种的表达矩阵
重命名基因:
- 去除物种前缀,恢复原始基因名称
- 例如:
GRCh38_CD79A→CD79A
分别整合:
- 人类细胞与单物种人类数据整合
- 小鼠细胞与单物种小鼠数据整合
示例代码:
# 读取混合物种数据
mixed_seurat <- Read10X("mixed_species_output/")
# 拆分人类和小鼠数据
human_genes <- grep("^GRCh38_", rownames(mixed_seurat), value = TRUE)
mouse_genes <- grep("^mm10_", rownames(mixed_seurat), value = TRUE)
# 提取人类细胞(根据gem_classification.csv)
human_cells <- classification$barcode[classification$call == "GRCh38"]
human_matrix <- mixed_seurat[human_genes, human_cells]
# 重命名基因(去除前缀)
rownames(human_matrix) <- gsub("^GRCh38_", "", rownames(human_matrix))
# 创建Seurat对象并与单物种数据整合
human_seurat <- CreateSeuratObject(human_matrix)
# ... 后续整合分析最后更新:2025 年 11 月