epiAneufinder 分析:基于 scATAC-seq 的非整倍体检测
背景介绍
染色体拷贝数变异(CNV)是肿瘤基因组不稳定性的典型标志,表现为染色体片段的大规模扩增或缺失。CNV 通过改变癌基因(如 MYC)或抑癌基因(如 TP53)的拷贝数,引发基因剂量效应,驱动肿瘤发生、进展与治疗抵抗。在单细胞分辨率下解析 CNV 对揭示肿瘤异质性尤为关键:依托恶性细胞特异的 CNV 图谱(如 7 号染色体扩增、10 号染色体缺失),可在复杂微环境中区分肿瘤细胞与非恶性细胞(如 T 细胞、成纤维细胞),进一步解析亚克隆结构及其表观调控差异。
相较于基于 scRNA-seq 的 CNV 推断,scATAC-seq 以基因组覆盖度为直接信号,不受转录程序与瞬时表达波动影响;在非转录区域也有更均匀的覆盖,因此对大片段与细粒度 CNV 的分辨率与稳健性更高。epiAneufinder 是专门面向 scATAC-seq 数据推断 CNV 的工具之一,利用基因组固定窗口的覆盖度计数作为 DNA 拷贝状态的代理信号,在单细胞层面识别染色体片段的扩增/缺失事件,从而实现恶性与非恶性细胞的鉴别与亚克隆精细刻画。
目的与意义
- 细胞级肿瘤鉴别:基于 CNV 图谱将恶性细胞与微环境细胞精准区分。
- 亚克隆异质性解析:重建拷贝数进化轨迹,理解克隆竞争与分化关系。
- 多组学联动解读:将 CNV 与染色质可及性、细胞注释/分群结果整合,阐明基因组结构改变对表观调控与细胞状态的影响。
- 临床转化价值:为预后分层、疗效评估与潜在靶点挖掘提供结构变异证据。
参数设置
请先挂载将要分析的云平台相关流程数据,挂载方式请参照 jupyter 使用教程
具体参数填写:
- meta:挂载的流程相关的 meta 文件,是流程相关 Seurat 对象里面的 meta.data ;
- species:物种信息,仅支持人和小鼠;
- celltype_col:细胞类型列名,选择要分析的细胞类型或者聚类对应的标签。假如想对注释好的细胞类型进行分析,则选择其对应的标签,例如 CellAnnotation;
- resolution_cluster:细胞类型,多选,选择要分析的细胞类型或者聚类结果,如 Monocyte 和 Macrophage 等;
- file_path:待分析样本的 fragment 文件,支持多个样本;
- SAMplenames:样本名,支持多个样本。
# Parameters
meta = "/PROJ2/FLOAT/advanced-analysis/test/2388-5504-5323-570556340787763834/meta.txt"
species = "human"
celltype_col = "CellAnnotation"
resolution_cluster = "wknn_res.0.5_d30_l2_50"
file_path=c("/PROJ2/FLOAT/advanced-analysis/test/2388-5504-6345-581728456711101050/joint14_A_fragments.tsv.gz","/PROJ2/FLOAT/advanced-analysis/test/2388-5504-6345-581728456711101050/joint22_A_fragments.tsv.gz")
samplenames = c("joint14","joint22")# 条件判断逻辑,加载物种信息
if (species == "human") {
genome <- "BSgenome.Hsapiens.UCSC.hg38"
blacklist <- "/PROJ2/FLOAT/shumeng/project/scATAC_learn/Signac/epiAneufinder/bin/hg38-blacklist.v2.bed"
} else if (species == "mouse") {
genome <- "BSgenome.Mmusculus.UCSC.mm10"
blacklist <- "/PROJ2/FLOAT/shumeng/project/scATAC_learn/Signac/epiAneufinder/bin/mm10-blacklist.v2.bed"
} else {
stop(paste0("Unsupported species '", species, "'. Supported: human, mouse."))
}环境准备
R 包加载
请选择 common_r 这个环境进行该整合教程的学习
options(warn = -1)
suppressWarnings({
suppressMessages({
Sys.setenv(R_DATATABLE_MMAP = "false")
library(data.table)
library(epiAneufinder,lib.loc="/PROJ2/FLOAT/shumeng/apps/miniconda3/envs/epiAneufinderR4.3.3/lib/R/library")
library(RColorBrewer)
library(Seurat)
library(ComplexHeatmap)
})
})
options(warn = 0)colors <- unique(c('#E5D2DD', '#53A85F', '#F1BB72', '#F3B1A0', '#D6E7A3', '#57C3F3', '#476D87',
'#E95C59', '#E59CC4', '#AB3282', '#23452F', '#BD956A', '#8C549C', '#585658',
'#9FA3A8', '#E0D4CA', '#5F3D69', '#C5DEBA', '#58A4C3', '#E4C755', '#F7F398',
'#AA9A59', '#E63863', '#E39A35', '#C1E6F3', '#6778AE', '#91D0BE', '#B53E2B',
'#712820', '#DCC1DD', '#CCE0F5', '#CCC9E6', '#625D9E', '#68A180', '#3A6963',
'#968175', "#6495ED", "#FFC1C1",'#f1ac9d','#f06966','#dee2d1','#6abe83','#39BAE8','#B9EDF8','#221a12',
'#b8d00a','#74828F','#96C0CE','#E95D22','#017890','#F1BB72', '#D6E7A3', '#57C3F3', '#476D87',
'#E95C59', '#E59CC4', '#AB3282', '#BD956A', '#8C549C', '#585658',
'#9FA3A8', '#E0D4CA', '#5F3D69', '#C5DEBA', '#58A4C3', '#E4C755', '#F7F398',
'#AA9A59', '#E63863', '#E39A35', '#C1E6F3', '#6778AE', '#91D0BE', '#B53E2B',
'#712820', '#DCC1DD', '#CCE0F5', '#CCC9E6'))Fragment 文件预处理
SeekArctools 或 cellranger arc/ATAC 生成的 fragments 文件包含所有条形码(barcode)的片段信息,其中仅有一部分为真实细胞。为节省计算资源并与后续细胞注释保持一致,进行 CNV 分析前需先对 fragments 进行过滤:仅保留通过质控并用于下游分析的“细胞白名单”条形码。建议依据已确定的细胞元数据/注释表(如 QC 合格且被纳入分析的 barcodes 列表)对子集化 fragments,从而避免将低质量条形码或非细胞背景噪声引入 CNV 推断。
meta <- read.table(meta,sep="\t",header=T) #读取挂载的meta文件,该文件包含挂载项目数据所有样本的barcode信息
Barcodes <- meta$barcodes #提取全部Barcode
#构建一个merged_filtered_fragments.tsv文件,方便后续依次处理各个样本fragment文件后将处理后的内容持续输出到这一个文件里
output_file <- "./merged_filtered_fragments.tsv"
# 在循环外先写入列名(如果不需要列名,可以跳过这步)
write.table(t(c("chr", "start", "end", "Barcode", "reads")),
file = output_file,
quote = FALSE,
row.names = FALSE,
col.names = FALSE,
sep = "\t")
# 修改循环
for(sample in samplenames){
file_path <- grep(pattern = sample, file_paths, value = TRUE) # 注意这里应该是 file_paths(复数)
frag <- fread(file_path, header=F)
# 修正这里的列名赋值语法错误
colnames(frag) <- c("chr", "start", "end", "Barcode", "reads") # 使用 <- 赋值
frag_filter <- frag[Barcode %in% Barcodes]
# 追加写入到同一个文件,append = TRUE 是关键参数
write.table(frag_filter,
file = output_file,
quote = FALSE,
row.names = FALSE,
col.names = FALSE, # 不写入列名,避免重复
sep = "\t",
append = TRUE) # 追加模式,而不是覆盖
}开始 CNV 分析
开始执行 epiAneufinder 的 CNV 推断流程。该流程将 scATAC-seq 片段数据转换为基因组固定窗口的覆盖度矩阵,通过噪声控制与事件识别,在单细胞层面解析染色体拷贝数变异。
执行步骤概述:
- 数据预处理:根据物种信息加载对应基因组与黑名单文件,设置窗口大小(100 kb)与过滤参数
- CNV 推断:调用 epiAneufinder 核心函数,执行分箱计数、噪声过滤与事件识别
- 结果输出:生成包含各窗口 CNV 状态的矩阵表格,支持后续可视化与细胞分群整合
关键参数说明:
- 窗口大小:默认 100 kb,平衡 CNV 分辨率与统计稳健性
- 细胞过滤:minFrags=20000,确保细胞覆盖度满足 CNV 推断要求
- 事件识别:k=4 聚类参数,minsize=1 最小事件长度,threshold_blacklist_bins=0.85 黑名单阈值
- 染色体排除:自动排除 chrX/Y/M,避免性别与线粒体差异干扰
options(future.globals.maxSize=100*1024^3)
invisible(capture.output(
suppressMessages(
epiAneufinder::epiAneufinder(input = output_file,#输入的fragment文件是上个环节中处理后并合并在一起的fragment文件
outdir = "./",
blacklist = blacklist,
windowSize = 1e5,
genome = genome,
exclude = c('chrX', 'chrY', 'chrM'),
reuse.existing = TRUE,
uq = 0.9,
lq = 0.1,
title_karyo = "Karyoplot",
ncores = 4,
minFrags = 20000,
minsize = 1,
k = 4,
threshold_blacklist_bins = 0.85,
minsizeCNV = 0
)
)
)
)处理结果文件
对上面 CNV 分析结果文件(../result/epiAneufinder_results/results_table.tsv)进行处理,方便可视化
meta <- meta[,c("barcodes",celltype_col,resolution_cluster)]
colnames(meta)[1] <- "cell"
colnames(meta)[2] <- "annot"
meta$cell <- paste0("cell-",meta$cell)
res_table <- read.table("./epiAneufinder_results/results_table.tsv", header = T,check.names = FALSE)
meta <- meta[which(meta$cell %in% colnames(res_table)),]
rownames(meta) <- meta$cell
res_table <- as.data.frame(res_table)
rownames(res_table) <- paste0(res_table$seq, ":", res_table$start, "-", res_table$end)
res_table <- res_table[,-c(1,2,3)]
res_table <- t(res_table)
chr <- colnames(res_table)
colann <- data.frame(
chromosome = stringr::str_split(chr, ":", simplify = TRUE)[,1],
position = stringr::str_split(chr, ":", simplify = TRUE)[,2]
)
rownames(colann) <- paste0(colann$chromosome,":",colann$position)
colann$chromosome <- gsub("chr", "", colann$chromosome)热图可视化
用热图可视化细胞拷贝数变异情况,copy number 值在 2 附近,表示正常的 2 倍体;copy number 在 1~2 之间,表示染色质存在缺失,越靠近 1,表示缺失越严重;copy number 在 2~3 之间表示染色质存在扩增, 越靠近 3 表示扩增越严重;热图左侧色带分别表示将细胞按照细胞类型和细胞聚类情况进行排列,每行表示一个细胞,每列表示一个染色体臂,热图的颜色表示 CNV 分数,颜色越红表示扩增越严重,颜色越蓝表示缺失越严重。
# 动态生成颜色(保持原代码不变)
n_clusters <- length(unique(meta[[resolution_cluster]]))
clusters_colors <- sample(colors, n_clusters, replace = FALSE)
n_celltypes <- length(unique(meta$annot))
cell_type_colors <- sample(colors, n_celltypes, replace = FALSE)
# 优化后的注释代码
ha <- rowAnnotation(
Cell_type = meta$annot,
Cluster = meta[[resolution_cluster]],
col = list(
Cluster = setNames(clusters_colors, unique(meta[[resolution_cluster]])),
Cell_type = setNames(cell_type_colors, unique(meta$annot))
),
annotation_label = list(
Cluster = "Cluster",
Cell_type = "Cell Type"
),
annotation_name_gp = gpar(fontsize = 8),
annotation_name_rot = 45,
annotation_legend_param = list(
Cluster = list(labels_gp = gpar(fontsize = 10)),
Cell_type = list(labels_gp = gpar(fontsize = 10))
),
width = unit(10, "cm")
)ht_opt$message = FALSE
p <- Heatmap(
res_table,
name = "Copy Number",
left_annotation = ha, # 取消注释并确保 ha 已定义
show_row_names = FALSE,
show_column_names = FALSE,
cluster_rows = TRUE,
cluster_columns = FALSE,
row_split = meta[,2],
row_title = "Cell Type",
column_split = factor(colann[,1],levels = c(1:22)), # 使用修正后的分组
column_gap = unit(1, "mm"), # 控制分割线间距
cluster_row_slices = FALSE,
use_raster = TRUE,
show_row_dend = FALSE,
border = FALSE
)options(repr.plot.height=10, repr.plot.width=15)
invisible(pdf("../result/cnv_heatmap_annotated.pdf", width = 15, height = 10))
invisible(draw(p))
invisible(dev.off())
draw(p)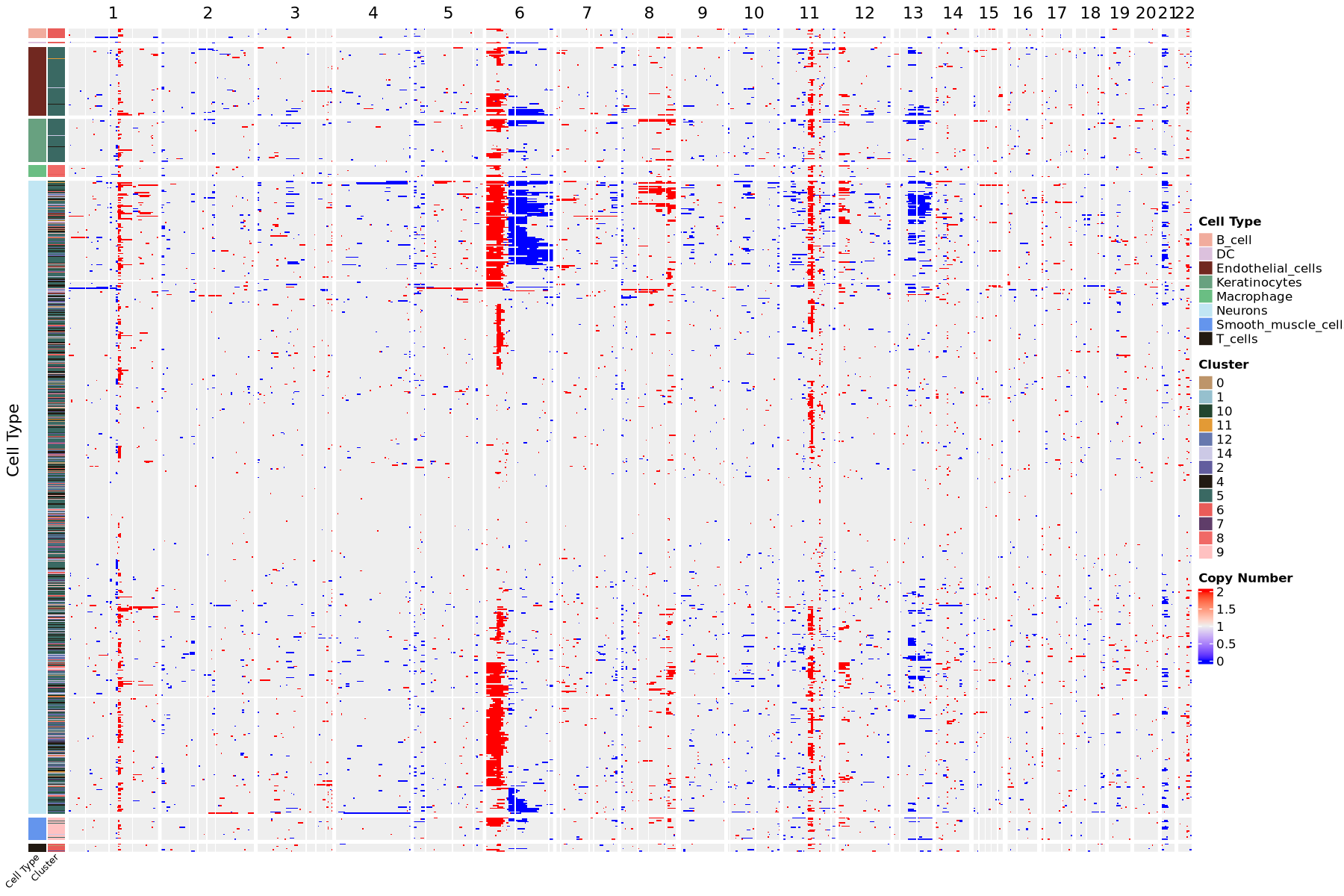
热图可视化
用热图可视化细胞拷贝数变异情况,copy number 值在 2 附近,表示正常的 2 倍体;copy number 在 1~2 之间,表示染色质存在缺失,越靠近 1,表示缺失越严重;copy number 在 2~3 之间表示染色质存在扩增, 越靠近 3 表示扩增越严重。
上面是热图的形式可视化染色体拷贝数变异情况,热图左侧色带分别表示将细胞按照细胞类型和细胞聚类情况进行排列,每行表示一个细胞,每列表示一个染色体臂,热图的颜色表示 CNV 分数,颜色越红表示扩增越严重,颜色越蓝表示缺失越严重。结果文件
├── ./epiAneufinder_results/karyo_annotated.pngcnv_plots
结果目录:目录下包括此项分析所涉及的所有图片以及表格等结果文件
文献案例解析
《The chromatin landscape of high-grade serous ovarian cancer metastasis identifies regulatory drivers in post-chemotherapy residual tumour cells》
在这项研究中,作者用 epiAneufinder 对 scATAC-seq 做拷贝数变异分析,用于区分肿瘤细胞和非肿瘤细胞
参考资料
[1] Nikolic A, Singhal D, Ellestad K, et al.Copy-scAT: Deconvoluting single-cell chromatin accessibility of genetic subclones in cancer[J].Science advances, 2021, 7(42):eabg6045.DOI: 10.1126/sciadv.abg6045.
附录
.txt :结果数据表格文件,文件以制表符(Tab)分隔。unix/Linux/Mac 用户使用 less 或 more 命令查看;windows 用户使用高级文本编辑器 Notepad++ 等查看,也可以用 Microsoft Excel 打开。
.pdf :结果图像文件,矢量图,可以放大和缩小而不失真,方便用户查看和编辑处理,可使用 Adobe Illustrator 进行图片编辑,用于文章发表等。
.rds : 包含基因活力 assay 的 Seurat 对象,需要在 R 环境中打开查看和用于进一步分析。