甲基化 + RNA 双组学: 差异分析和功能富集分析流程
模块简介
本模块整合了单细胞甲基化差异分析和功能富集分析两个核心流程,旨在从单细胞多组学数据中识别差异甲基化区域/基因、差异表达基因,并进一步解析其生物学功能。
分析内容概览
第一部分:差异分析
- DMB (Differentially Methylated Bins):差异甲基化区间分析,识别不同细胞类型间甲基化水平存在显著差异的基因组区域(20 kb 窗口)。
- DEG (Differentially Expressed Genes):差异表达基因分析,识别不同细胞类型间表达水平存在显著差异的基因。
- DMG (Differentially Methylated Genes):差异甲基化基因分析,识别不同细胞类型间甲基化水平存在显著差异的基因(基因区域 ±2 kb)。
- 多组学关联分析:整合转录组和甲基化数据,通过维恩图和 Circos 图揭示表观遗传修饰与基因表达之间的调控关系。
第二部分:功能富集分析
- GO 富集分析:基于基因本体(Gene Ontology)数据库,识别差异基因富集的生物学过程(BP)、分子功能(MF)和细胞组成(CC)。
- KEGG 富集分析:基于 KEGG 通路数据库,识别差异基因富集的生物学通路。
技术特点
- 统计方法:使用 Wilcoxon Rank-Sum test 进行差异分析,多重比较校正(FDR)控制假阳性率。
- 自动化流程:集成数据预处理、差异分析、功能富集和可视化于一体。
- 多组学整合:同时分析转录组和甲基化数据,揭示表观遗传调控机制。
💡 Note
本分析流程适用于已完成细胞类型注释的单细胞多组学数据,需要同时具备转录组(RNA)和甲基化(Methylation)数据。
输入文件准备
必需文件
本模块需要以下输入文件:
| 文件类型 | 文件格式 | 必需列/属性 | 说明 |
|---|---|---|---|
| 转录组数据 | *.h5ad | obs['celltype'], var['gene_ids'] | 已注释细胞类型的转录组数据,需包含细胞类型信息和基因 ID |
| 甲基化数据 | *.h5ad | obs['celltype'], obs['Sample'] | 已注释细胞类型的甲基化数据,需包含细胞类型和样本信息 |
| MCDS 数据 | *.mcds | chrom20k, geneslop2k | 多样本 MCDS 数据,包含 20 kb 窗口和基因区域 ±2 kb 的甲基化信息 |
文件结构要求
目录结构示例:
data/
├── AY1768874914782
│ └── methylation
│ └── demoWTJW969-task-1
│ └── WTJW969
│ ├── allcools_generate_datasets
│ │ └── WTJW969.mcds # 甲基化数据
│ ├── filtered_feature_bc_matrix # 转录组表达矩阵
│ └── split_bams
│ ├── WTJW969_cells.csv
│ └── filtered_barcode_reads_counts.csv
└── AY1768876253533
└── methylation
└── demo4WTJW880-task-1
└── WTJW880
├── allcools_generate_datasets
│ └── WTJW880.mcds # 甲基化数据
├── filtered_feature_bc_matrix # 转录组表达矩阵
└── split_bams
├── WTJW880_cells.csv
└── filtered_barcode_reads_counts.csv数据预处理要求
- 完整注释:转录组和甲基化数据必须包含完整的细胞类型注释(
celltype列) - QC 完成:转录组数据需完成标准化处理
- MCDS 准备:MCDS 数据需包含
chrom20k和geneslop2k两个维度
⚠️ 重要提示
缺失注释信息将无法进行后续分析。请确保所有必需列都存在且格式正确。
import os
import re
import glob
from ALLCools.mcds import MCDS
from ALLCools.clustering import tsne, significant_pc_test, log_scale, lsi, binarize_matrix, filter_regions, cluster_enriched_features, ConsensusClustering, Dendrogram, get_pc_centers
from ALLCools.clustering.doublets import MethylScrublet
from ALLCools.plot import *
import scanpy as sc
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
from matplotlib.lines import Line2D
import warnings
import xarray as xr
from ALLCools.clustering import one_vs_rest_dmg
import pybedtools
from scipy import sparse
from pycirclize import Circos
from pycirclize.utils import load_eukaryote_example_dataset
# 功能富集分析相关包
import sys
import subprocess
from pathlib import Path
import gseapy as gp
from gseapy import barplot, dotplot
from IPython.display import display, HTML# 差异分析参数
samples = ["WTJW969", "WTJW880"]
sample_path_config = {
"WTJW969": {"top_dir": "AY1768874914782", "demo_dir": "demoWTJW969-task-1"},
"WTJW880": {"top_dir": "AY1768876253533", "demo_dir": "demo4WTJW880-task-1"}
}
anno_col = 'celltype' # 细胞类型注释列名
# 功能富集分析参数
species = "human" # 物种:'human' 或 'mouse'
deg_file = "DEG.csv" # 差异表达基因文件路径
dmg_file = "DMG.csv" # 差异甲基化基因文件路径
output_dir = "./enrichment_results/" # 富集分析结果输出目录
## 画图色板
my_palette = [
"#66C2A5", "#FC8D62", "#8DA0CB", "#E78AC3",
"#A6D854", "#FFD92F", "#E5C494", "#B3B3B3",
"#8DD3C7", "#BEBADA", "#FB8072", "#80B1D3",
"#FDB462", "#B3DE69", "#FCCDE5", "#BC80BD",
"#CCEBC5", "#FFED6F", "#A6CEE3", "#FB9A99"
]数据加载
数据加载是分析流程的第一步,需要确保转录组和甲基化数据都已正确注释,并且细胞标签(barcode)保持一致。
读取转录组和甲基化数据
读取已注释细胞类型的转录组和甲基化 h5ad 文件。这两个文件是后续所有分析的基础。
注意事项:
- 检查细胞类型注释是否完整,缺失注释的细胞将无法参与后续分析。
- 建议在加载数据后检查细胞数量和细胞类型分布,确保数据质量。
# 读取转录组数据
# 使用scanpy读取h5ad格式的转录组数据
# h5ad是AnnData格式,是单细胞分析的标准数据格式
adata_rna = sc.read_h5ad("adata_rna.h5ad")
print(f"转录组数据: {adata_rna.n_obs} 个细胞, {adata_rna.n_vars} 个基因")
# 读取甲基化数据
# 甲基化数据也使用h5ad格式存储,包含细胞类型注释和样本信息
adata_met = sc.read_h5ad("adata_met.h5ad")
print(f"甲基化数据: {adata_met.n_obs} 个细胞")
# 检查细胞类型注释
# 验证转录组和甲基化数据中的细胞类型是否一致
# 确保两个数据集的细胞类型注释列存在且包含相同的细胞类型
print(f"\n转录组细胞类型: {adata_rna.obs[anno_col].unique()}")
print(f"甲基化细胞类型: {adata_met.obs[anno_col].unique()}")甲基化数据: 2226 个细胞
转录组细胞类型: ['Memory CD4+ T', 'CD8+ T', 'Navie CD4+ T', 'CD14+ Mono', 'FCGR3A+ Mono', 'NK', 'DC', 'B']
Categories (8, object): ['B', 'CD8+ T', 'CD14+ Mono', 'DC', 'FCGR3A+ Mono', 'Memory CD4+ T', 'NK', 'Navie CD4+ T']
甲基化细胞类型: ['CD8+ T', 'NK', 'B', 'CD14+ Mono', 'Memory CD4+ T', 'DC', 'FCGR3A+ Mono', 'Navie CD4+ T']
Categories (8, object): ['B', 'CD14+ Mono', 'CD8+ T', 'DC', 'FCGR3A+ Mono', 'Memory CD4+ T', 'NK', 'Navie CD4+ T']
差异甲基化区间 (DMB) 分析
差异甲基化区间 (Differentially Methylated Bins, DMBs) 指的是在不同细胞群之间甲基化水平存在显著差异的基因组区域。识别 DMBs 有助于发现细胞特异性的表观遗传调控元件。
DMB 分析的意义:
- 发现调控元件:DMB 通常位于基因调控区域(启动子、增强子等),可能影响基因表达。
- 细胞类型特异性:不同细胞类型具有不同的甲基化模式,DMB 反映了这种特异性。
- 表观遗传标记:DMB 可以作为细胞类型的表观遗传标记,用于细胞类型鉴定。
分析策略:
- 使用 20 kb 固定窗口(chrom20k)进行全基因组扫描,平衡分辨率和统计功效。
- 采用「一对多」(one-vs-rest)策略,识别每种细胞类型相对于其他所有细胞类型的特异性甲基化区域。
- 结合统计显著性(FDR 校正 P 值)、差异倍数(Fold Change)和区分能力(AUROC)进行综合筛选。
在 chrom20k 甲基化水平上合并多样本 MCDS
首先需要在 20 kb 窗口(chrom20k)水平上合并多个样本的 MCDS 数据。这是 DMB 分析的基础步骤。
合并流程:
- 读取各样本 MCDS:从每个样本目录中读取对应的 MCDS 文件,仅保留在甲基化数据中存在的细胞 barcode
- 重命名细胞标签:为避免不同样本间的细胞 barcode 冲突,为每个样本的细胞添加样本标识后缀
- 合并数据:使用
xarray.concat将所有样本的 MCDS 数据沿细胞维度合并 - 添加注释信息:为合并后的数据添加样本信息(
Sample)和细胞类型信息(anno_col),便于后续分组分析
关键参数:
obs_dim='cell':指定细胞维度名称。var_dim='chrom20k':指定 20 kb 窗口维度名称。use_obs=keep_barcodes:仅使用存在于甲基化数据中的细胞,确保数据一致性。
warnings.filterwarnings('ignore')
# 合并多样本MCDS数据(chrom20k水平)
# 此步骤将多个样本的MCDS数据合并为一个统一的数据集,用于后续的差异分析
mcds_list = [] # 存储每个样本的MCDS对象
cell_number = [] # 记录每个细胞所属的样本
# 遍历每个样本,读取对应的MCDS数据
for i in samples:
# 提取该样本在甲基化数据中的细胞barcode(去除后缀,匹配MCDS中的barcode格式)
keep_barcodes = [ re.sub('\\-.*','',b) for b in adata_met.obs[adata_met.obs["Sample"] == i].index ]
# 打开MCDS文件,仅保留存在于甲基化数据中的细胞
# obs_dim='cell': 指定细胞维度名称
# var_dim="chrom20k": 指定20kb窗口维度名称
mcds = MCDS.open(os.path.join('../../','data', sample_path_config[i]['top_dir'], 'methylation', sample_path_config[i]['demo_dir'], i, 'allcools_generate_datasets', f'{i}.mcds'),
obs_dim = 'cell', var_dim = "chrom20k", use_obs = keep_barcodes)
# 如果有多个样本,为每个细胞的barcode添加样本标识后缀,避免barcode冲突
suffix = samples.index(i)
if len(samples) > 1:
mcds = mcds.assign_coords(cell=[ f'{i}-{suffix}' for i in mcds.cell.values ])
mcds_list.append(mcds)
cell_number += [i]*len(mcds.cell.values) # 记录每个细胞所属的样本
# 合并所有样本的MCDS数据
# 使用xarray.concat沿细胞维度合并
combined = xr.concat(mcds_list, dim='cell')
# 添加样本信息坐标
combined = combined.assign_coords(Sample=('cell', cell_number))
# 添加细胞类型注释坐标
combined = combined.assign_coords(anno_col=('cell', adata_met.obs[anno_col]))
# 设置MCDS对象的维度属性
combined.obs_dim = 'cell'
combined.var_dim = "chrom20k"
print(f"合并后的MCDS数据: {len(combined.cell)} 个细胞, {len(combined.chrom20k)} 个20kb窗口")特征筛选流程
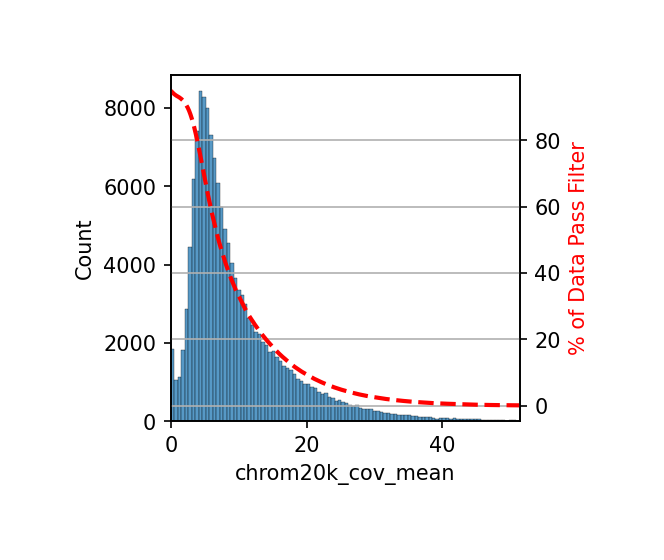
根据覆盖度均值进行特征筛选,保留高质量的特征区域。这是确保 DMB 分析质量的关键步骤。
特征筛选的重要性:
- 提高数据质量:低覆盖度的区域可能由于测序深度不足而产生噪声,影响差异分析的准确性。
- 减少假阳性:过滤掉覆盖度不足的区域可以降低假阳性率,提高结果的可靠性。
- 提高计算效率:减少需要分析的特征数量,加快分析速度。
筛选流程:
- 计算覆盖度均值:
add_feature_cov_mean()函数计算每个 20 kb 窗口在所有细胞中的平均覆盖度 - 设置阈值:根据数据质量设置
min_cov参数(默认值为 1),表示至少需要 1 个细胞在该区域有覆盖 - 过滤特征:
filter_feature_by_cov_mean()函数移除平均覆盖度低于阈值的区域
参数调整建议:
- 高质量数据:可以使用
min_cov=1或更高值,确保所有保留的区域都有足够的覆盖度。 - 低质量数据:如果保留的特征过少,可以适当降低
min_cov(如 0.5),但需注意可能引入噪声。 - 平衡考虑:建议先使用默认值,如果保留的特征数量过少,再考虑降低阈值。
# 计算特征覆盖度均值
# 覆盖度均值反映每个20kb窗口在所有细胞中的平均覆盖情况
# 用于后续的特征筛选,过滤掉覆盖度不足的低质量区域
# 设置matplotlib绘图参数
plt.rcParams['figure.dpi'] = 150
plt.rcParams['figure.figsize'] = (3,3)
# 计算每个20kb窗口在所有细胞中的平均覆盖度
# 结果存储在 combined.coords['chrom20k_cov_mean'] 中
combined.add_feature_cov_mean()
print("特征覆盖度均值已计算完成")特征覆盖度均值已计算完成
# 根据覆盖度筛选特征
# 过滤掉平均覆盖度低于阈值的20kb窗口,提高数据质量
min_cov = 1 # 最小覆盖度阈值,表示至少需要1个细胞在该区域有覆盖
# 如果保留的特征过少,可以适当降低此值(如0.5),但可能引入噪声
# 筛选特征:移除平均覆盖度 < min_cov 的窗口
combined.filter_feature_by_cov_mean(min_cov=min_cov)
print(f"覆盖度筛选完成,保留的特征区域数量: {len(combined.chrom20k)}")After cov mean filter: 144048 chrom20k 93.3%
覆盖度筛选完成,保留的特征区域数量: 154423
计算甲基化水平并保存 MCDS 数据
计算每个 20 kb 窗口的甲基化水平(甲基化分数),并保存合并后的 MCDS 数据供后续分析使用。
# 计算甲基化分数(fraction)
# 甲基化分数 = 甲基化位点数 / (甲基化位点数 + 非甲基化位点数)
# 反映每个20kb窗口的甲基化水平(0-1范围)
# normalize_per_cell=True: 对每个细胞进行归一化,消除细胞间测序深度差异
# clip_norm_value=10: 将归一化值限制在10以内,避免极端值影响
combined.add_mc_frac(normalize_per_cell=True, clip_norm_value=10)
# 仅保留甲基化分数数据,减少内存占用
combined = combined[['chrom20k_da_frac']]
# 转换为float32类型,节省内存
combined['chrom20k_da_frac'] = combined['chrom20k_da_frac'].astype('float32')
# 保存后可以在后续分析中直接加载,避免重复计算
combined.write_dataset(output_path = 'merge_chrom20k.mcds')
print("MCDS数据已保存到 merge_chrom20k.mcds")Saving chunk 0: 0 - 1000
Saving chunk 1: 1000 - 2000
Saving chunk 2: 2000 - 2226
MCDS数据已保存到 merge_chrom20k.mcds
差异甲基化区间分析
使用 one_vs_rest_dmg 函数进行差异甲基化区间分析,识别每种细胞类型相对于其他细胞类型的特异性甲基化区域。
设置差异分析参数
min_cov = 1 # 最小覆盖度(已在前面步骤中筛选) obs_dim = 'cell' # 观测维度(细胞) var_dim = 'chrom20k' # 变量维度(20kb窗口) mc_type = 'CGN' # 甲基化类型:CGN表示CG位点的甲基化 top_n = 1000 # 每个细胞类型保留的Top N个差异区间 auroc_cutoff = 0.1 # AUROC阈值,用于过滤区分度较弱的差异区间 adj_p_cutoff = 0.05 # 校正P值阈值(FDR < 0.05视为显著) fc_cutoff = np.inf # Fold Change阈值(设为无穷大表示不限制) max_cluster_cells = 5000 # 每个细胞类型用于分析的最大细胞数(超过会随机采样) max_other_fold = 5 # 其他细胞类型的最大倍数(用于平衡样本量) cpu = 16 # 并行计算的CPU核心数(实际使用中设置为1以避免阻塞)
执行差异甲基化区间分析
one_vs_rest_dmg函数采用"一对多"策略,识别每种细胞类型相对于其他所有细胞类型的特异性甲基化区域
使用Wilcoxon Rank-Sum test进行统计检验,并进行FDR校正
dmb_table = one_vs_rest_dmg(adata_met.obs, # 细胞元数据,包含细胞类型注释 group=anno_col, # 细胞类型注释列名 mcds_paths='merge_chrom20k.mcds', # MCDS数据路径 obs_dim=obs_dim, var_dim=var_dim, mc_type=mc_type, top_n=top_n, adj_p_cutoff=adj_p_cutoff, fc_cutoff=fc_cutoff, auroc_cutoff=auroc_cutoff, max_cluster_cells=max_cluster_cells, max_other_fold=max_other_fold, cpu=1) # 设置为1以避免阻塞
print(f"DMB分析完成,共识别 {len(dmb_table)} 个差异甲基化区间")
保存结果到HDF5格式(高效存储)
dmb_table.to_hdf(f'merge_{var_dim}.OneVsRestDMB.hdf', key='data')
cmd = f'/jp_envs/envs/seeksoulmethyl/bin/python ./run_dmg.py adata_met.h5ad celltype chrom20k merge_chrom20k.mcds'
os.system(cmd)warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
Calculating cluster B DMGs.
Calculating cluster NK DMGs.
Calculating cluster DC DMGs.
Calculating cluster FCGR3A+ Mono DMGs.
Calculating cluster Memory CD4+ T DMGs.
Calculating cluster CD14+ Mono DMGs.
Calculating cluster Navie CD4+ T DMGs.
Calculating cluster CD8+ T DMGs.
DC Finished.
FCGR3A+ Mono Finished.
B Finished.
CD14+ Mono Finished.
CD8+ T Finished.
NK Finished.
Memory CD4+ T Finished.
Navie CD4+ T Finished.
0
# 读取DMB分析结果
dmb_table = pd.read_hdf('merge_chrom20k.OneVsRestDMB.hdf')
# 添加bin的基因组坐标信息
bin_info = pd.DataFrame({
'bin_id': combined["chrom20k"].values,
'chrom': combined['chrom20k_chrom'].values,
'start': combined['chrom20k_start'].values,
'end': combined['chrom20k_end'].values}
)
# 合并结果
dmb_table = dmb_table.merge(bin_info, left_index = True, right_on = 'bin_id')
# 按 chrom 和 bin_id 排序,表格 index 从 0 开始
dmb_table = dmb_table.sort_values(['chrom', 'bin_id']).reset_index(drop=True)
print(f"DMB结果已合并,共 {len(dmb_table)} 个差异甲基化区间")
print(dmb_table.head(n=10))DMB 关键指标说明:
- pvals_adj:Wilcoxon 检验的多重比较校正后 P 值,反映该 bin 在「该 cluster vs 其他细胞」比较中的显著性
- fc:该 cluster 内细胞的平均甲基化分数与簇外细胞的平均甲基化分数之比(in/out fold change)。fc < 1 表示该 cluster 相对其他细胞低甲基化
- AUROC:该 bin 在区分「目标 cluster 内细胞」与「其他细胞」上的 AUC,经过对称化处理为 abs(AUC-0.5)+0.5,越接近 1 区分度越强
- cluster:目标 cluster
- bin_id:bin 的 ID
- chrom, start, end:bin 所在的染色体号和基因组坐标
差异表达基因 (DEG) 分析
差异表达基因 (Differentially Expressed Genes, DEGs) 指的是在不同细胞群中表达水平存在显著差异的基因。使用 Wilcoxon Rank-Sum test 识别每种细胞类型的特异性标记基因(Marker Genes)。
DEG 分析的意义:
- 细胞类型标记:DEG 可以作为细胞类型的特异性标记基因,用于细胞类型鉴定和注释。
- 功能特征:高表达的 DEG 反映了该细胞类型的主要功能特征和生物学过程。
- 多组学整合:DEG 与 DMG 的联合分析可以揭示表观遗传修饰对基因表达的调控作用。
统计方法:
- Wilcoxon Rank-Sum test:非参数检验方法,不假设数据分布,适用于单细胞数据的特点(稀疏性、零膨胀)。
- 多重比较校正:使用 Benjamini-Hochberg 方法进行 FDR 校正,控制假发现率。
- 效应量评估:使用 Log2 Fold Change 评估表达差异的幅度,通常以 |Log2FC| > 0.25 作为筛选标准。
分析流程:
- 分组比较:对每个细胞类型,将其作为目标组,其他所有细胞类型作为对照组
- 统计检验:对每个基因进行 Wilcoxon 检验,计算显著性 P 值
- 多重校正:对所有基因的 P 值进行 FDR 校正,得到校正后 P 值(
pvals_adj) - 结果排序:根据统计量(scores)或校正 P 值对结果进行排序,识别最显著的差异基因
# 使用scanpy进行差异表达基因分析
# rank_genes_groups函数对每个细胞类型进行"一对多"比较
# method='wilcoxon': 使用Wilcoxon Rank-Sum test(非参数检验,适用于单细胞数据)
# groupby=anno_col: 按细胞类型分组
# pts=True: 计算基因在目标组和参考组中的表达比例
sc.tl.rank_genes_groups(adata_rna, method = 'wilcoxon', groupby = anno_col, pts = True)
# 提取所有细胞类型的差异表达基因结果
# group=None: 提取所有细胞类型的结果
DEG = sc.get.rank_genes_groups_df(adata_rna, group = None)
# 将列名 group 改为 cluster,与后续分析一致
DEG = DEG.rename(columns={'group': 'cluster'})
# 保存结果到CSV文件
DEG.to_csv('DEG.csv', index = False)
print(f"DEG分析完成,共识别 {len(DEG)} 个差异表达基因")
print(DEG.head(n=10))DEG 关键指标说明:
- cluster:目标细胞簇(Cluster)
- names:基因名称
- scores:Z-score 统计量,绝对值越大表示差异越显著
- logfoldchanges:对数折叠变化(Log2 Fold Change)。正值表示在目标簇中高表达,负值表示低表达
- pvals:原始 P 值
- pvals_adj:经 Benjamini-Hochberg 校正后的 P 值(FDR)。通常以 < 0.05 作为显著性阈值
- pct_nz_group:目标簇中表达该基因的细胞比例
- pct_nz_reference:其他簇中表达该基因的细胞比例
差异甲基化基因 (DMG) 分析
差异甲基化基因 (Differentially Methylated Genes, DMGs) 指的是在不同细胞类型间甲基化水平存在显著差异的基因。分析基于基因区域±2kb(geneslop2k)的甲基化水平。
在 geneslop2k 甲基化水平上合并多样本 MCDS
# 合并多样本MCDS数据(geneslop2k水平)
# 此步骤与chrom20k合并类似,但使用基因区域±2kb(geneslop2k)维度
# geneslop2k维度包含每个基因及其上下游2kb区域的甲基化信息
warnings.filterwarnings('ignore')
mcds_list = [] # 存储每个样本的MCDS对象
cell_number = [] # 记录每个细胞所属的样本
# 遍历每个样本,读取对应的MCDS数据
for i in samples:
# 提取该样本在甲基化数据中的细胞barcode(去除后缀)
keep_barcodes = [ re.sub('\\-.*','',b) for b in adata_met.obs[adata_met.obs["Sample"] == i].index ]
# 打开MCDS文件,使用geneslop2k维度(基因区域±2kb)
# var_dim='geneslop2k': 指定基因区域维度名称
mcds = MCDS.open(os.path.join('../../','data', sample_path_config[i]['top_dir'], 'methylation', sample_path_config[i]['demo_dir'], i, 'allcools_generate_datasets', f'{i}.mcds'),
obs_dim = 'cell', var_dim = 'geneslop2k', use_obs = keep_barcodes)
# 如果有多个样本,为每个细胞的barcode添加样本标识后缀
suffix = samples.index(i)
if len(samples) > 1:
mcds = mcds.assign_coords(cell=[ f'{i}-{suffix}' for i in mcds.cell.values ])
mcds_list.append(mcds)
cell_number += [i]*len(mcds.cell.values)
# 合并所有样本的MCDS数据
combined_gene = xr.concat(mcds_list, dim="cell")
# 添加样本信息坐标
combined_gene = combined_gene.assign_coords(Sample=("cell", cell_number))
# 设置MCDS对象的维度属性
combined_gene.obs_dim = 'cell'
combined_gene.var_dim = 'geneslop2k'
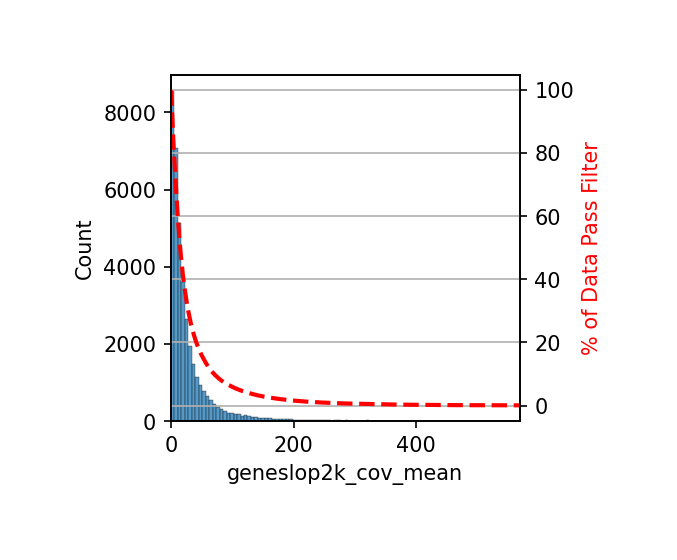
print(f"合并后的MCDS数据: {len(combined_gene.cell)} 个细胞, {len(combined_gene.geneslop2k)} 个基因区域")# 计算特征覆盖度均值并筛选
# 与chrom20k分析类似,对基因区域进行覆盖度筛选,保留高质量区域
plt.rcParams['figure.dpi'] = 150
plt.rcParams['figure.figsize'] = (3,3)
# 计算每个基因区域在所有细胞中的平均覆盖度
combined_gene.add_feature_cov_mean()
# 筛选特征:移除平均覆盖度 < 1 的基因区域
combined_gene.filter_feature_by_cov_mean(min_cov=1)
print(f"覆盖度筛选完成,保留的基因区域数量: {len(combined_gene.geneslop2k)}")Before cov mean filter: 38569 geneslop2k
After cov mean filter: 37508 geneslop2k 97.2%
覆盖度筛选完成,保留的基因区域数量: 38569
# 计算真实甲基化分数(用于后续可视化)
combined_gene.add_mc_frac(normalize_per_cell=False, clip_norm_value=None, da_suffix = 'true_frac')
geneslop2k_da_true_frac = combined_gene['geneslop2k_da_true_frac']
# 计算标准化甲基化分数(用于差异分析)
combined_gene.add_mc_frac(normalize_per_cell=True, clip_norm_value=10)
combined_gene = combined_gene[[f'geneslop2k_da_frac']]
combined_gene[f'geneslop2k_da_frac'] = combined_gene[f'geneslop2k_da_frac'].astype('float32')
# 保存合并后的MCDS数据
combined_gene.write_dataset(output_path = 'merge_geneslop2k.mcds')
print("MCDS数据已保存到 merge_geneslop2k.mcds")Saving chunk 0: 0 - 1000
Saving chunk 1: 1000 - 2000
Saving chunk 2: 2000 - 2226
MCDS数据已保存到 merge_geneslop2k.mcds
差异甲基化基因分析
使用 one_vs_rest_dmg 函数对每个细胞类型进行差异甲基化基因(DMG)分析。该函数采用「一对多」(one-vs-rest)策略,识别每种细胞类型相对于其他所有细胞类型的特异性甲基化基因。
分析方法:
- 统计检验:Wilcoxon Rank-Sum test(Mann-Whitney U test),用于比较目标细胞类型与其他细胞类型在基因区域甲基化水平上的差异。
- 多重比较校正:Benjamini-Hochberg 方法(FDR 校正),控制假发现率。
- 筛选标准:结合校正 P 值(
adj_p_cutoff)、Fold Change(fc_cutoff)和 AUROC(auroc_cutoff)进行综合筛选。
设置差异分析参数
min_cov = 1 obs_dim = 'cell' var_dim = 'geneslop2k' mc_type = 'CGN' top_n = 1000 auroc_cutoff = 0.1 adj_p_cutoff = 0.05 fc_cutoff = np.inf max_cluster_cells = 5000 max_other_fold = 5 cpu = 16
执行差异甲基化基因分析
dmg_table = one_vs_rest_dmg(adata_met.obs, group=anno_col, mcds_paths='merge_geneslop2k.mcds', obs_dim='cell', var_dim='geneslop2k', mc_type=mc_type, top_n=top_n, adj_p_cutoff=adj_p_cutoff, fc_cutoff=fc_cutoff, auroc_cutoff=auroc_cutoff, max_cluster_cells=max_cluster_cells, max_other_fold=max_other_fold, cpu=1)
保存结果
dmg_table.to_hdf(f'merge_{anno_col}.OneVsRestDMG.hdf', key='data') print(f"DMG分析完成,共识别 {len(dmg_table)} 个差异甲基化基因")
# 如果使用外部脚本运行,可以使用以下命令
cmd = f'/jp_envs/envs/seeksoulmethyl/bin/python ./run_dmg.py adata_met.h5ad celltype geneslop2k merge_geneslop2k.mcds'
os.system(cmd)warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/jp_envs/envs/seeksoulmethyl/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
Calculating cluster DC DMGs.
Calculating cluster FCGR3A+ Mono DMGs.
Calculating cluster CD14+ Mono DMGs.
Calculating cluster Navie CD4+ T DMGs.
Calculating cluster CD8+ T DMGs.
Calculating cluster NK DMGs.
Calculating cluster B DMGs.
Calculating cluster Memory CD4+ T DMGs.
DC Finished.
B Finished.
CD8+ T Finished.
CD14+ Mono Finished.
FCGR3A+ Mono Finished.
NK Finished.
Memory CD4+ T Finished.
Navie CD4+ T Finished.
0
# 读取DMG分析结果
dmg_table = pd.read_hdf('merge_celltype.OneVsRestDMG.hdf', key='data')
# 保存为CSV格式
dmg_table.reset_index().to_csv('DMG.csv', index = False)
print(f"DMG结果已保存,共 {len(dmg_table)} 个差异甲基化基因")
dmg_table = dmg_table.reset_index()
dmg_table['names'] = [ re.sub('.*_','',i) for i in dmg_table['names'] ]
print(dmg_table.head())DMG 关键指标说明:
- names:基因名称
- pvals_adj:Wilcoxon 检验的多重比较校正后 P 值,反映该 gene 在「该 cluster vs 其他细胞」比较中的显著性
- fc:该 cluster 内细胞的平均甲基化分数与簇外细胞的平均甲基化分数之比(in/out fold change)。fc < 1 表示该 cluster 相对其他细胞低甲基化
- AUROC:该 gene 在区分「目标 cluster 内细胞」与「其他细胞」上的 AUC,经过对称化处理为 abs(AUC-0.5)+0.5,越接近 1 区分度越强
- cluster:目标 cluster
差异甲基化基因可视化
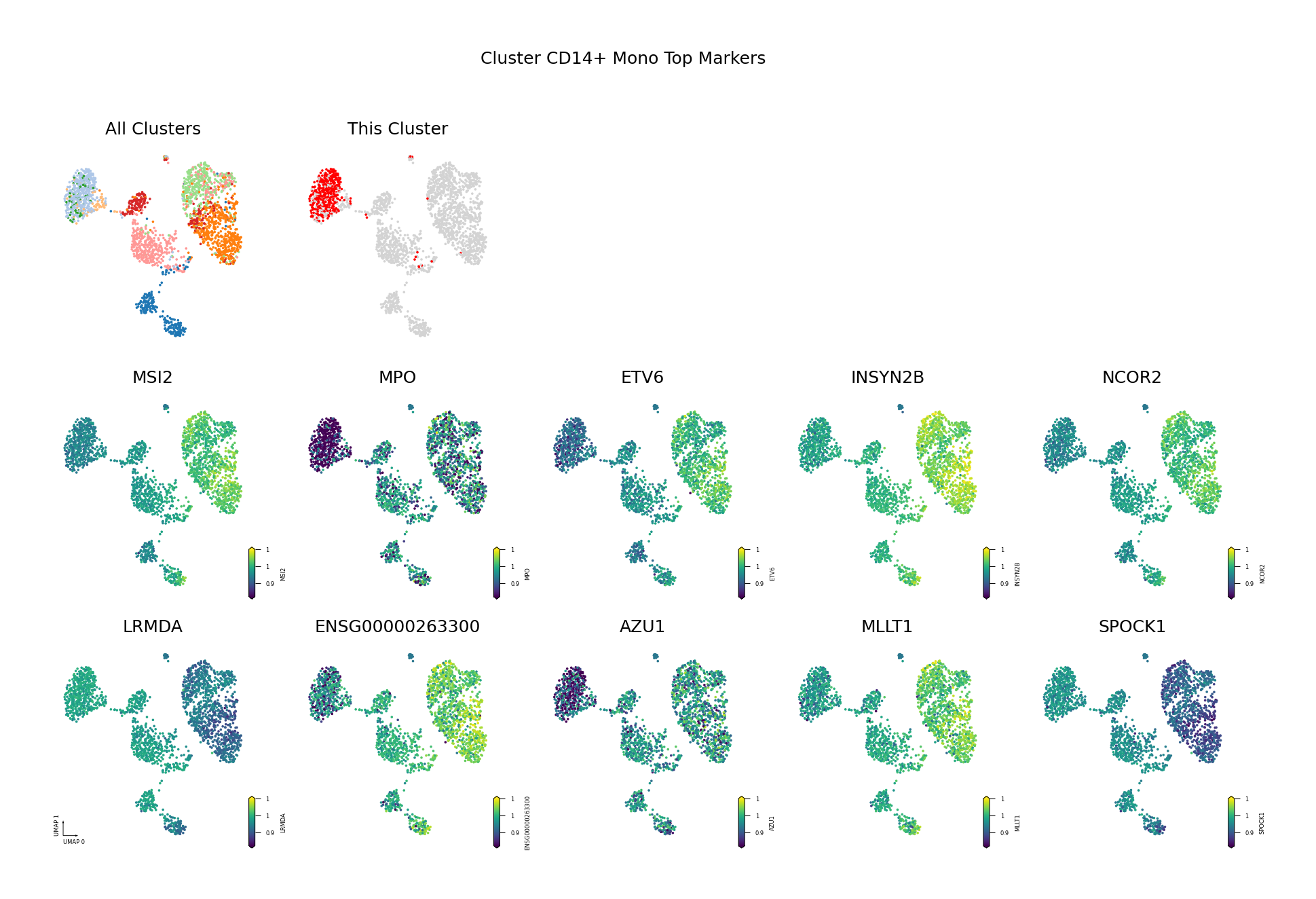
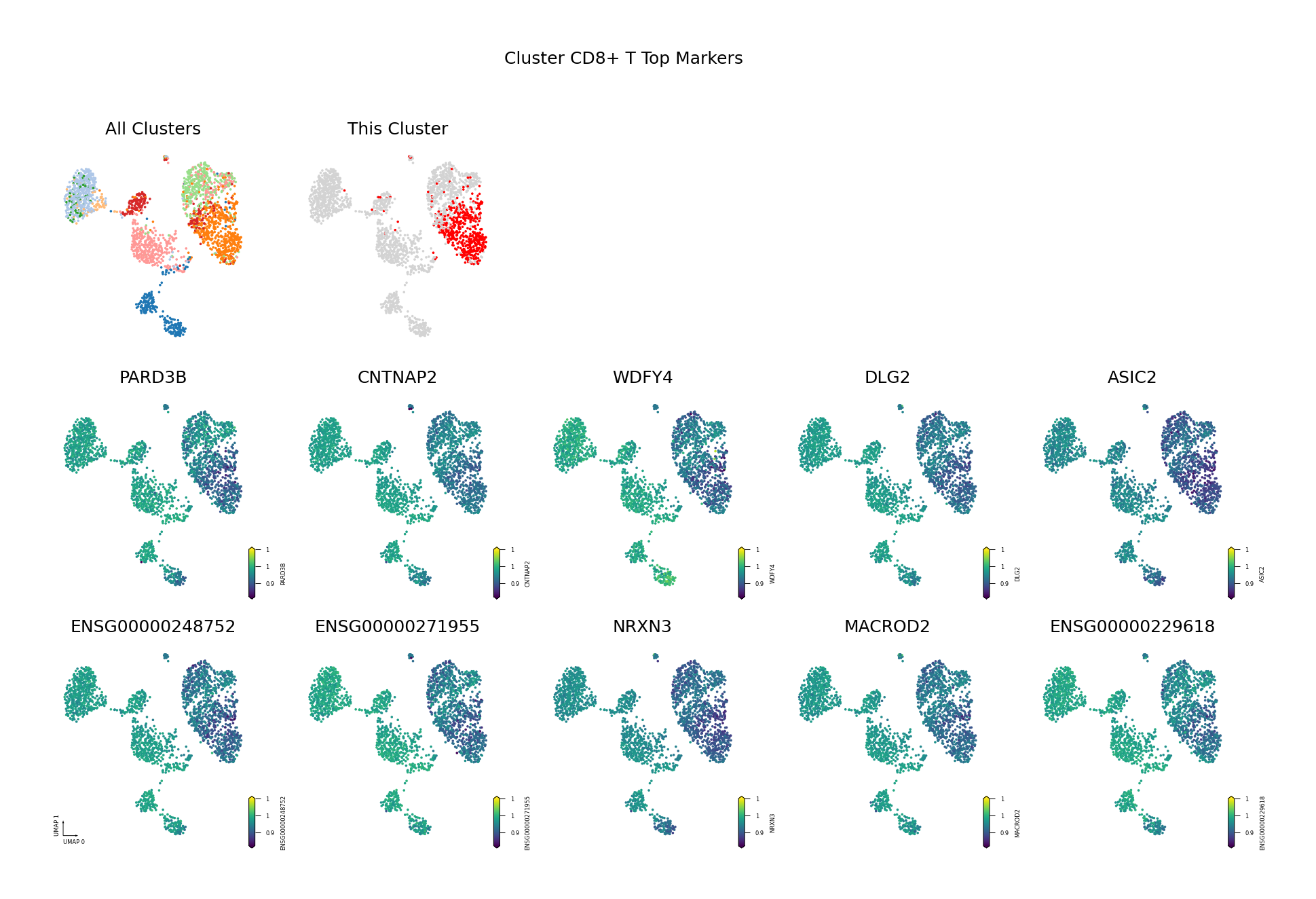
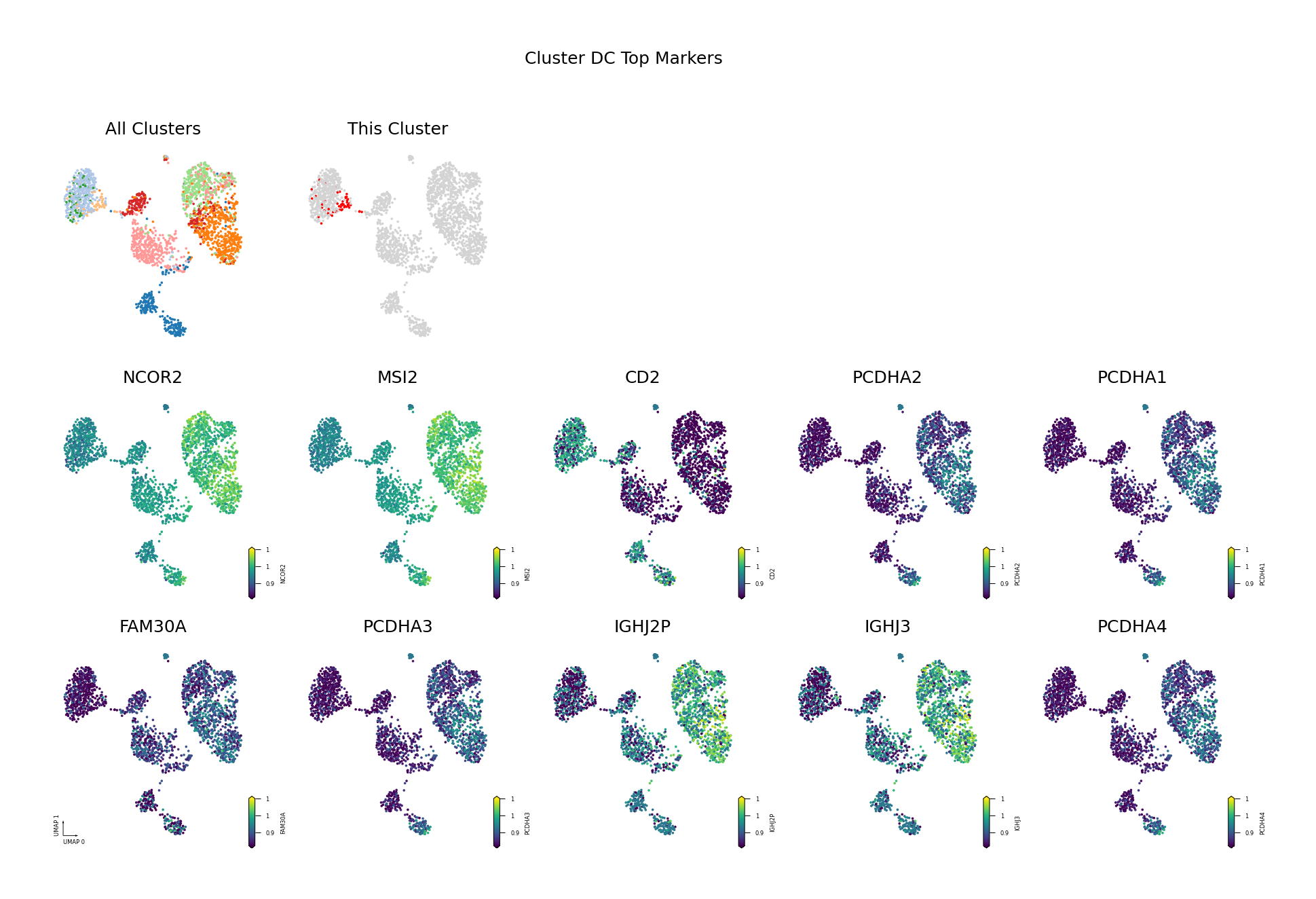
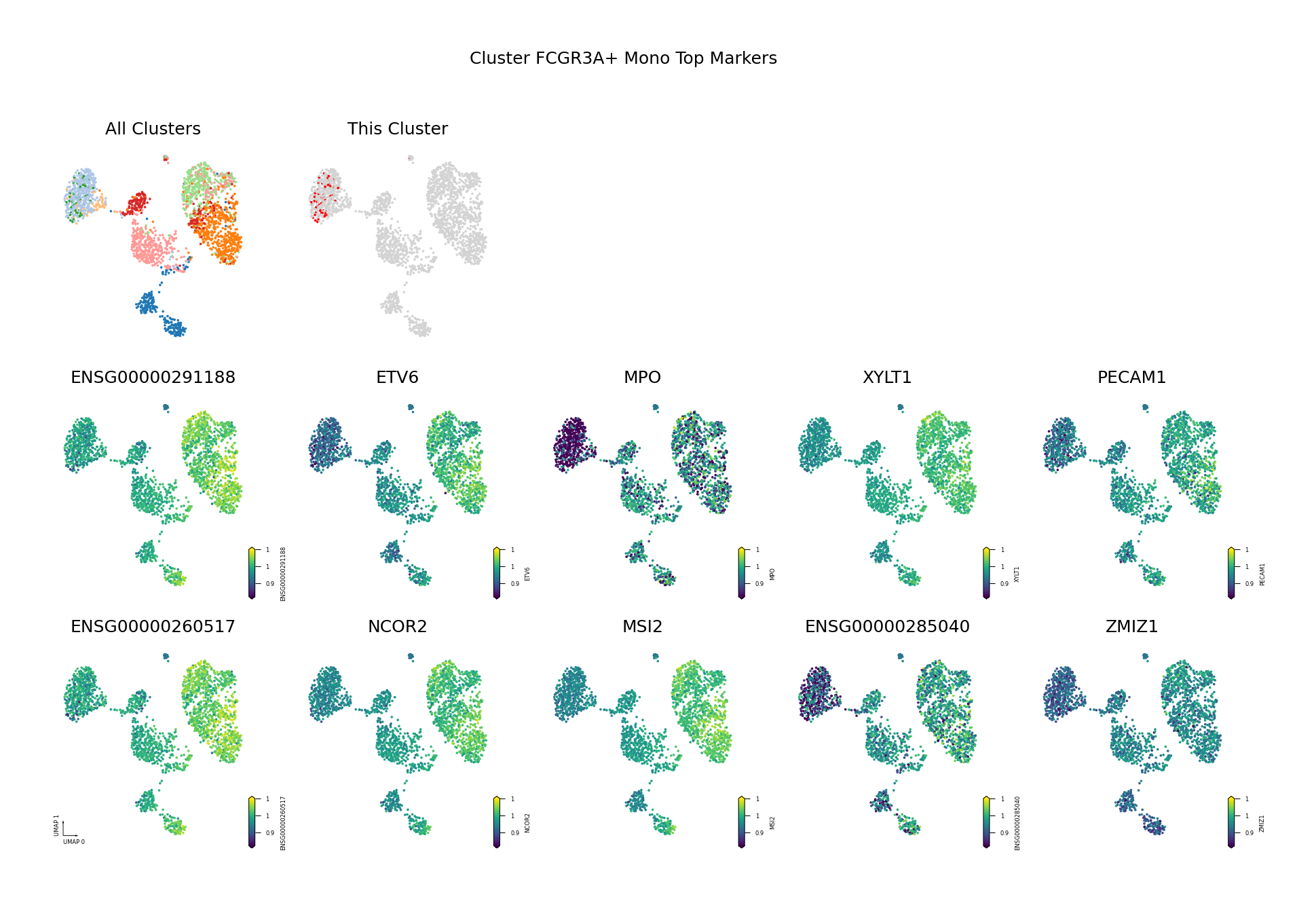
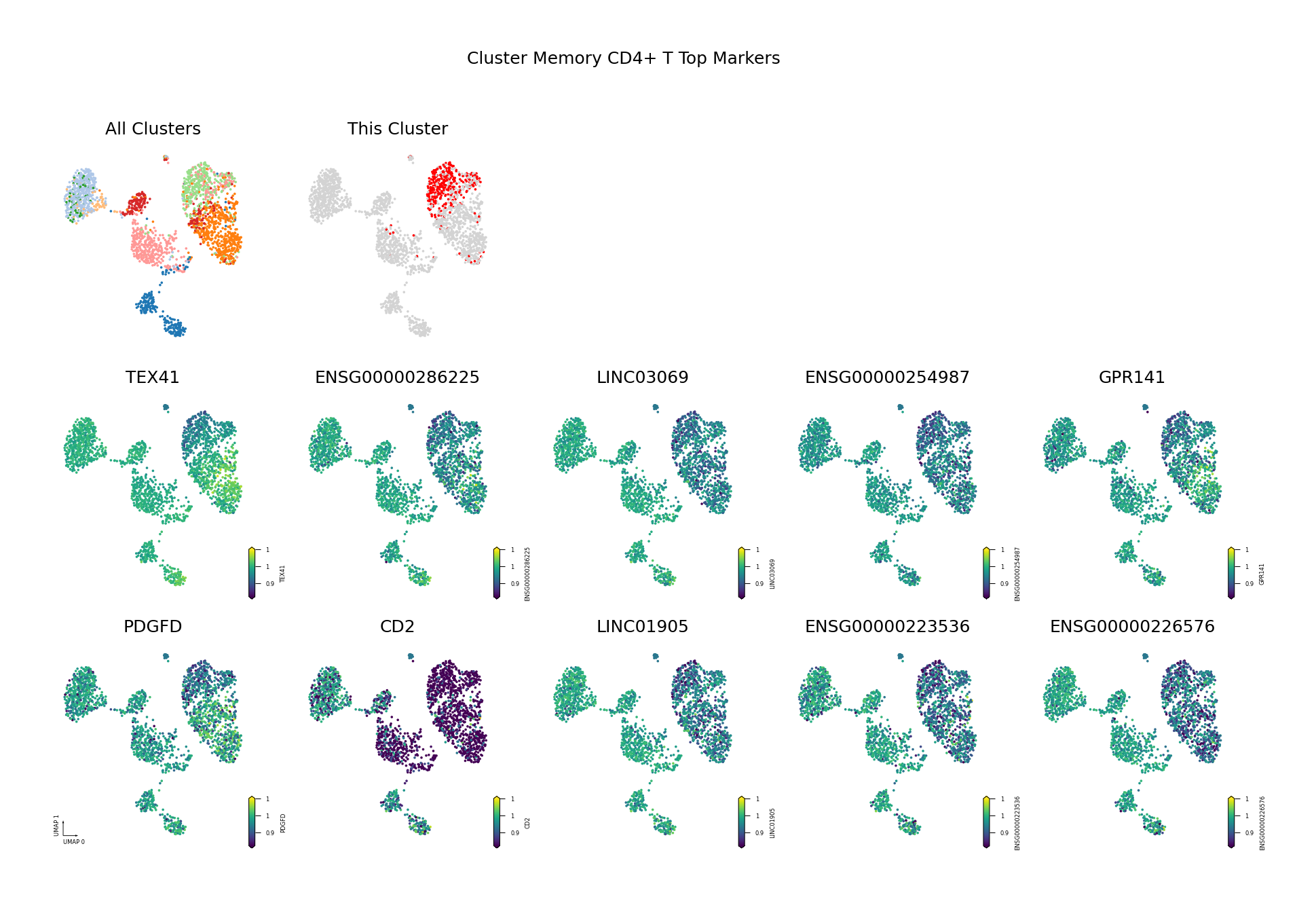
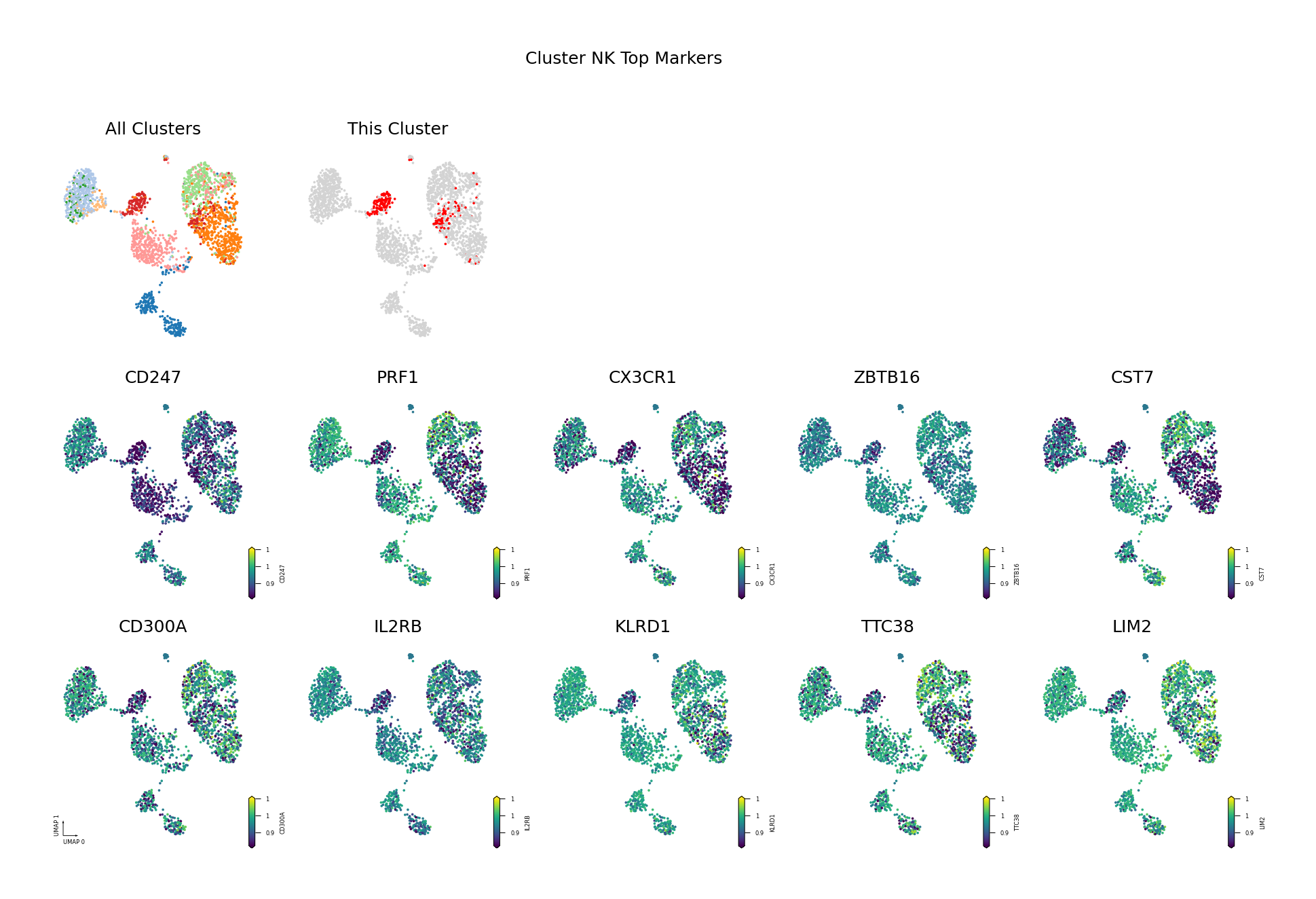
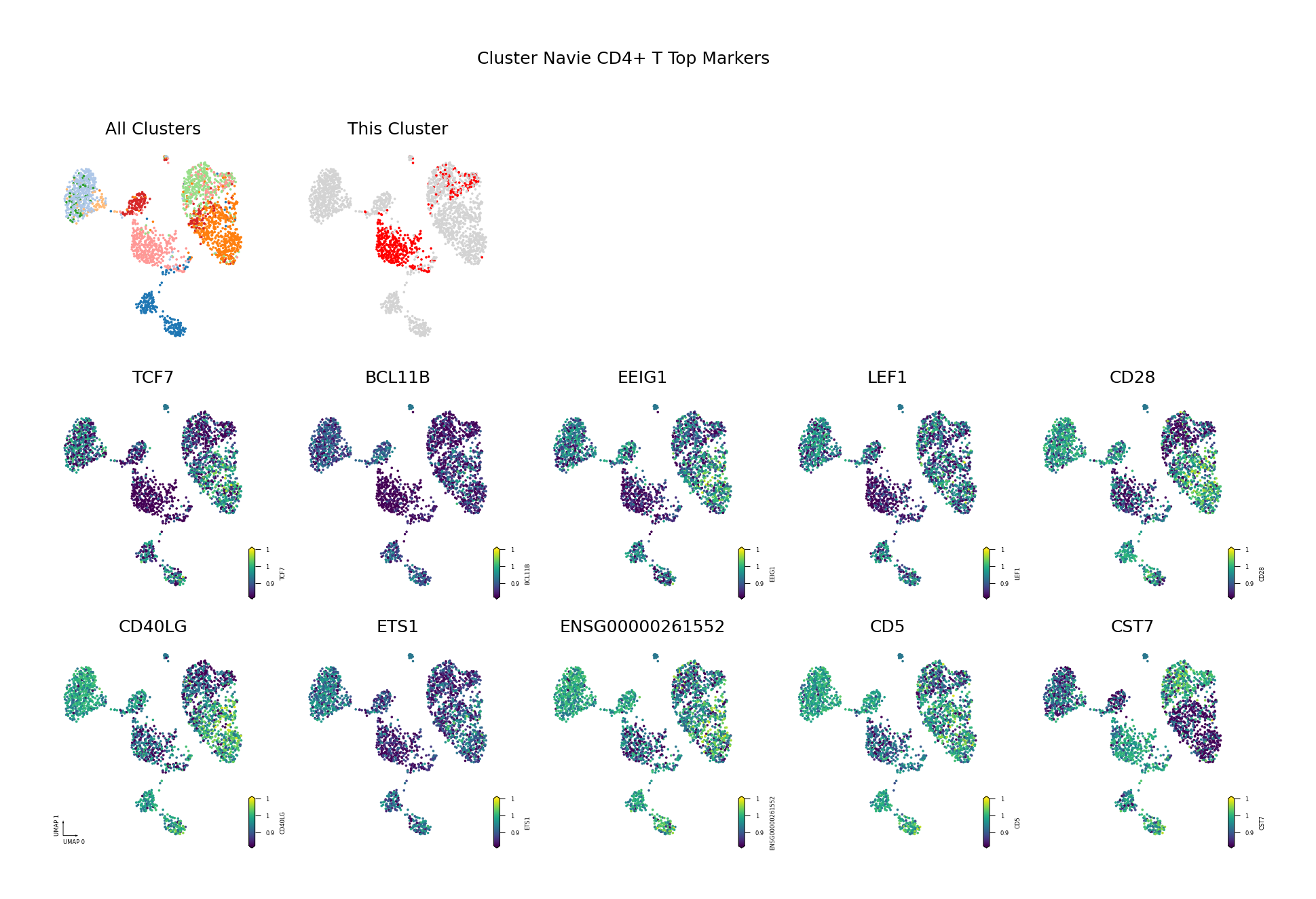
为了直观展示识别出的差异甲基化基因,我们将筛选出的 Top DMG(基于 AUROC 排序,仅展示前 10 个)的甲基化水平投射到 UMAP 空间中。
可视化目的:
- 验证差异模式:通过 UMAP 可视化验证识别出的 DMG 是否确实在目标细胞类型中表现出特异的甲基化模式。
- 空间分布:观察 DMG 的甲基化水平在 UMAP 空间中的分布,是否与细胞类型边界一致。
- 发现模式:识别具有相似甲基化模式的基因,可能参与相同的生物学过程。
筛选标准:AUROC > 0.6 且(Fold Change < 0.9 或 > 1.1)
- AUROC > 0.6:确保基因具有良好的区分能力,能够有效区分目标细胞类型与其他细胞类型。
- Fold Change 阈值:筛选出具有明显甲基化差异的基因。
- FC < 0.9:低甲基化基因(在目标细胞类型中甲基化水平较低)。
- FC > 1.1:高甲基化基因(在目标细胞类型中甲基化水平较高)。
图注:本图展示每个细胞类型中 Top 10 差异甲基化基因(DMG)的甲基化水平在 UMAP 降维空间中的分布。
- 第一行左侧图:所有细胞类型的 UMAP 图,不同颜色代表不同细胞类型,展示整体细胞分布与边界
- 第一行右侧图:目标细胞类型的 UMAP 图,红色高亮目标细胞类型,灰色为其他细胞类型
- 后续子图:各 Top 10 DMG 的甲基化水平在 UMAP 空间中的分布,一子图一基因
- 横轴/纵轴:UMAP_1 与 UMAP_2,表示降维后的两个主维度
- 颜色:表示甲基化水平(fraction),越深(红/黄)越高,越浅(蓝/紫)越低
- 点:每个点为一个细胞,位置表示细胞间相似性
def plot_cluster_and_genes(cluster, cell_meta, cluster_col, genes_data,
coord_base='umap', ncols=5, axes_size=3, dpi=150, hue_norm=(0.67, 1.5)):
"""绘制细胞类型和Top DMGs的UMAP可视化图"""
ncols = max(2, ncols)
nrows = 1 + (genes_data.shape[1] - 1) // ncols + 1
# figure
fig = plt.figure(figsize=(ncols * axes_size, nrows * axes_size), dpi=dpi)
gs = fig.add_gridspec(nrows=nrows, ncols=ncols)
# cluster axes
ax = fig.add_subplot(gs[0, 0])
categorical_scatter(data=cell_meta,
ax=ax,
coord_base=coord_base,
axis_format=None,
hue=cluster_col,
palette='tab20')
ax.set_title('All Clusters')
ax = fig.add_subplot(gs[0, 1])
categorical_scatter(data=cell_meta,
ax=ax,
coord_base=coord_base,
hue=cell_meta.obs[cluster_col] == cluster,
axis_format=None,
palette={
True: 'red',
False: 'lightgray'
})
ax.set_title('This Cluster')
# gene axes
for i, (gene, data) in enumerate(genes_data.items()):
col = i % ncols
row = i // ncols + 1
ax = fig.add_subplot(gs[row, col])
if ax.get_subplotspec().is_first_col() and ax.get_subplotspec().is_last_row():
axis = 'tiny'
else:
axis = None
continuous_scatter(ax=ax,
data=cell_meta,
hue=data,
axis_format=axis,
hue_norm=hue_norm,
coord_base=coord_base)
ax.set_title(f'{data.name}')
fig.suptitle(f'Cluster {cluster} Top Markers')
return fig
# 准备基因注释信息
dmg_table = pd.read_csv("DMG.csv",index_col=0)
clusters = sorted(set(dmg_table['cluster']))
geneslop2k_bed = pd.DataFrame({
'geneslop2k_id': combined_gene['geneslop2k'].values,
'geneslop2k_gene_ids': [ re.sub('_.*','',i) for i in combined_gene['geneslop2k'].values ],
'geneslop2k_name': [ re.sub('.*_','',i) for i in combined_gene['geneslop2k'].values],
'geneslop2k_chrom': combined_gene['geneslop2k_chrom'].values,
'geneslop2k_start': combined_gene['geneslop2k_start'].values,
'geneslop2k_end':combined_gene['geneslop2k_end'].values,
})
gene_name_to_gene_id = {v.geneslop2k_name:v.geneslop2k_id for idx, v in geneslop2k_bed.iterrows()}
geneslop2k_bed = geneslop2k_bed.set_index('geneslop2k_id')
# 筛选显著的DMG
dmg_table_plot = dmg_table[(dmg_table.AUROC > 0.6) & ((dmg_table.fc < 0.9) | (dmg_table.fc > 1.1))]
# 降采样以节省内存
downsample = 30000
if downsample and (adata_met.n_obs > downsample):
use_cells = adata_met.obs.sample(downsample, random_state=0).index
adata_met_subset = adata_met[adata_met.obs_names.isin(use_cells), :].copy()
else:
use_cells = adata_met.obs_names
adata_met_subset = adata_met
# 加载基因甲基化数据
gene_frac_da = MCDS.open(f'merge_geneslop2k.mcds',use_obs=use_cells)[f'geneslop2k_da_frac']
gene_frac_da = gene_frac_da.load()
# 为每个cluster绘制Top DMGs
for cluster in clusters:
genes = dmg_table_plot[dmg_table_plot['cluster'] == cluster].sort_values('AUROC', ascending=False)[:10]
if genes.shape[0] < 1:
continue
else:
genes_data = gene_frac_da.sel(geneslop2k=genes.index).to_pandas()
genes_data.columns = genes_data.columns.map(geneslop2k_bed['geneslop2k_name'])
fig = plot_cluster_and_genes(cluster=cluster,
cell_meta=adata_met,
cluster_col=anno_col,
genes_data=genes_data,
coord_base='umap',
ncols=5,
axes_size=3,
dpi=150)
# 可选:保存图片
# fig.savefig(f'figures/{cluster.replace(" ","_")}.TopDMG.pdf',dpi=300, bbox_inches='tight')
# fig.savefig(f'figures/{cluster.replace(" ","_")}.TopDMG.png', dpi=300, bbox_inches='tight')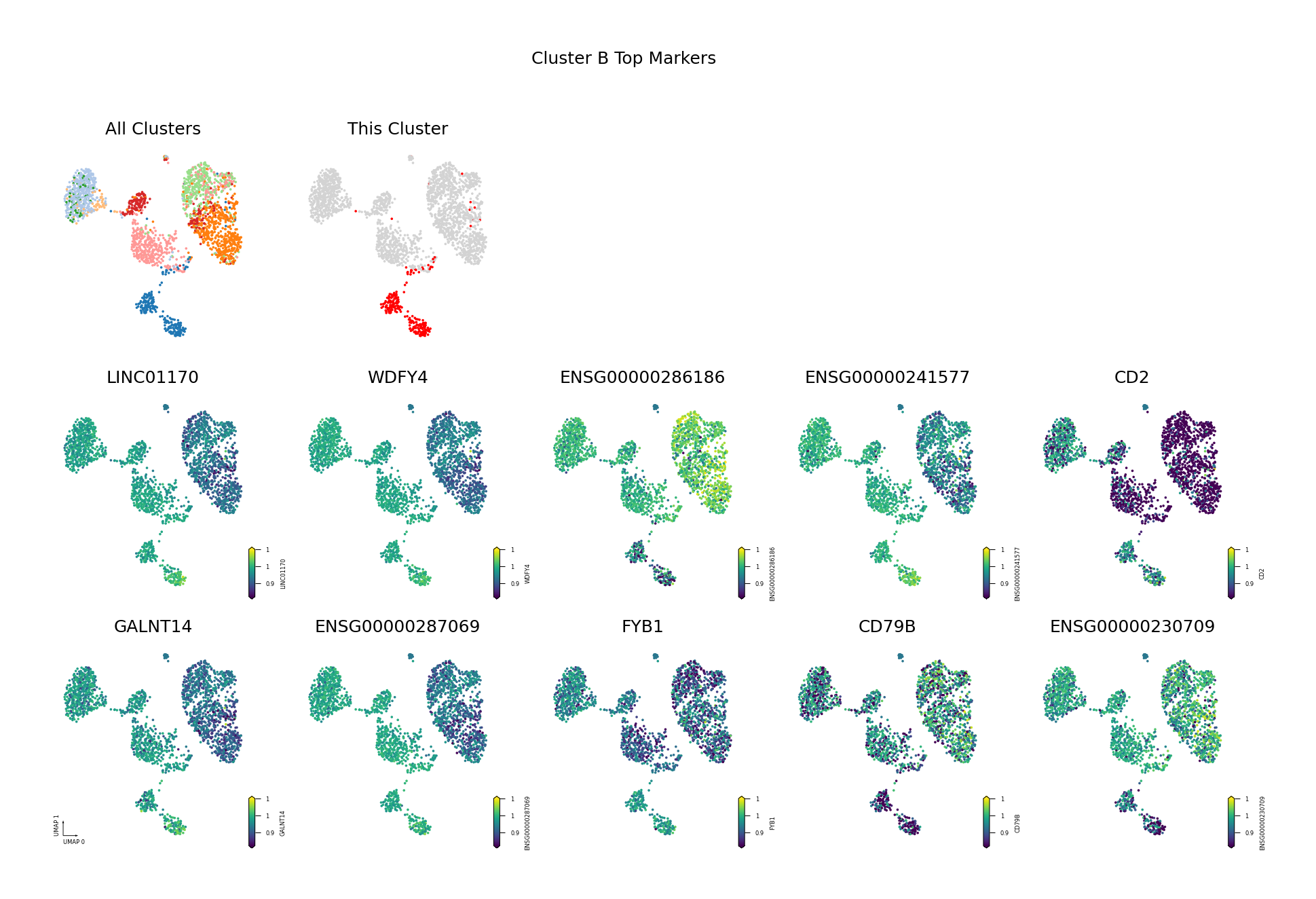
多组学关联分析
整合转录组(RNA)和甲基化(Methylation)数据,揭示表观遗传修饰与基因表达之间的调控关系。通常情况下,基因启动子区域的高甲基化会抑制基因表达(负相关),但基因主体(Gene Body)的甲基化与表达的关系可能更为复杂。
多组学关联分析的意义:
- 揭示调控机制:通过整合转录组和甲基化数据,可以识别表观遗传修饰对基因表达的调控作用。
- 发现关键基因:同时具有差异表达和差异甲基化的基因可能是细胞类型分化和功能的关键调控因子。
- 理解生物学过程:多组学整合有助于理解细胞类型特异性功能的表观遗传基础。
甲基化与表达的关联模式:
- 启动子高甲基化 → 低表达:这是最常见的负相关模式,启动子区域的高甲基化通常抑制基因转录。
- 启动子低甲基化 → 高表达:启动子区域的低甲基化通常促进基因转录。
- 基因体甲基化:基因主体(Gene Body)的甲基化与表达的关系较为复杂,可能因基因类型而异。
- 增强子甲基化:增强子区域的甲基化变化可能影响远距离基因的表达。
分析内容:
- 重叠分析:识别同时具有 DEG 和 DMG 状态的基因,这些基因可能是表观遗传调控的关键靶点
- 方向性分析:分析表达变化方向(高表达/低表达)与甲基化变化方向(高甲基化/低甲基化)的关联模式
- 可视化展示:通过维恩图和 Circos 图直观展示多组学数据的关联关系
# 准备DMG数据
dmg_table['met_status'] = ['hypo_met' if i < 1 else 'hyper_met' for i in dmg_table['fc']]
dmg_table['gene_ids'] = [ re.sub('_.*','',i) for i in dmg_table.index ]
dmg_table['gene_names'] = [ re.sub('.*_','',i) for i in dmg_table.index ]
# 准备DEG数据
deg_table = DEG.copy()
deg_table = deg_table.merge(adata_rna.var[['gene_ids']], left_on = 'names', right_index = True, how = 'left')
deg_table['cluster_gene'] = deg_table['cluster'].str.cat(deg_table['gene_ids'],sep = '_')
deg_table = deg_table[(deg_table.pvals_adj < 0.05) & (abs(deg_table.logfoldchanges) > 0.25)]
deg_table['exp_status'] = np.where(deg_table['logfoldchanges'] > 0, 'high_exp',
np.where(deg_table['logfoldchanges'] < 0, 'low_exp', 'neutral'))
# 合并DEG和DMG数据
rename_col = {}
for i in dmg_table.columns:
rename_col[i] = f'met_{i}'
dmg_table_renamed = dmg_table.rename(columns = rename_col)
dmg_table_renamed['cluster_gene'] = dmg_table_renamed['met_cluster'].str.cat(dmg_table_renamed['met_gene_ids'], sep = '_')
merge_deg_dmg = deg_table.merge(dmg_table_renamed, how = 'outer', left_on = 'cluster_gene', right_on = 'cluster_gene')
conditions = [
(merge_deg_dmg['logfoldchanges'].notna() & merge_deg_dmg['met_fc'].isna()),
(merge_deg_dmg['logfoldchanges'].isna() & merge_deg_dmg['met_fc'].notna()),
(merge_deg_dmg['logfoldchanges'].notna() & merge_deg_dmg['met_fc'].notna()),
(merge_deg_dmg['logfoldchanges'].isna() & merge_deg_dmg['met_fc'].isna()),
]
choices = ['DEG', 'DMG', 'Both', 'Both']
merge_deg_dmg['status'] = np.select(conditions, choices, default='Both')
# 保存同时具有DEG和DMG的基因
merge_deg_dmg[merge_deg_dmg['status'] == 'Both'].to_csv('DEG_DMG.csv', index = False)
print(f"多组学关联分析完成,共 {len(merge_deg_dmg[merge_deg_dmg['status'] == 'Both'])} 个同时具有DEG和DMG的基因")
# 添加基因组坐标信息
dmg_table_renamed = dmg_table_renamed.merge(geneslop2k_bed, right_on = 'geneslop2k_gene_ids', left_on = 'met_gene_ids', how = 'left')
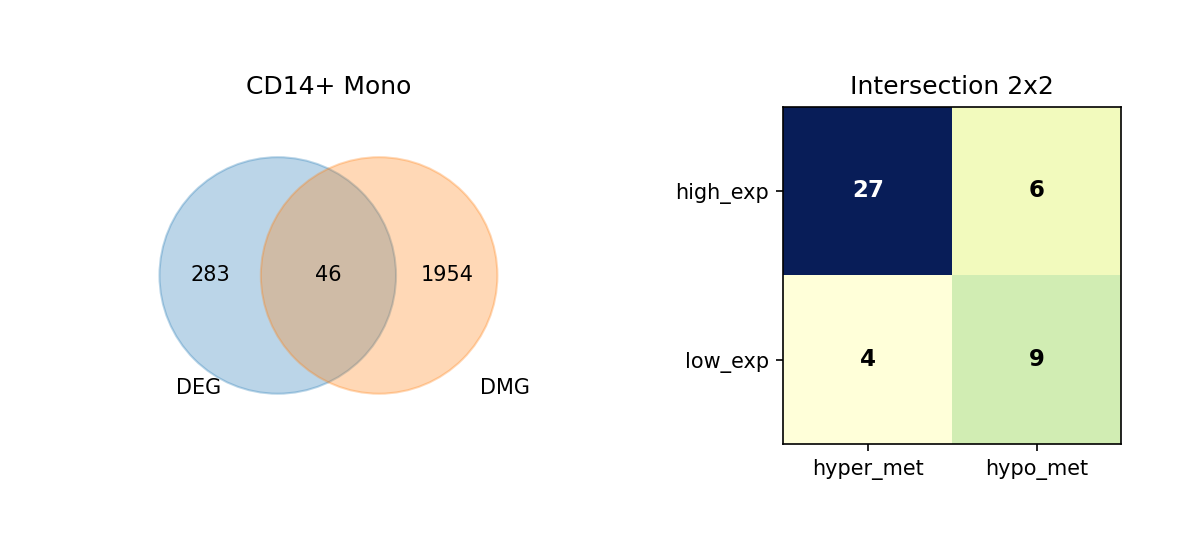
deg_table = deg_table.merge(geneslop2k_bed, right_on = 'geneslop2k_gene_ids', left_on = 'gene_ids', how = 'left')差异基因与差异甲基化基因的重叠分析(Venn Diagrams)
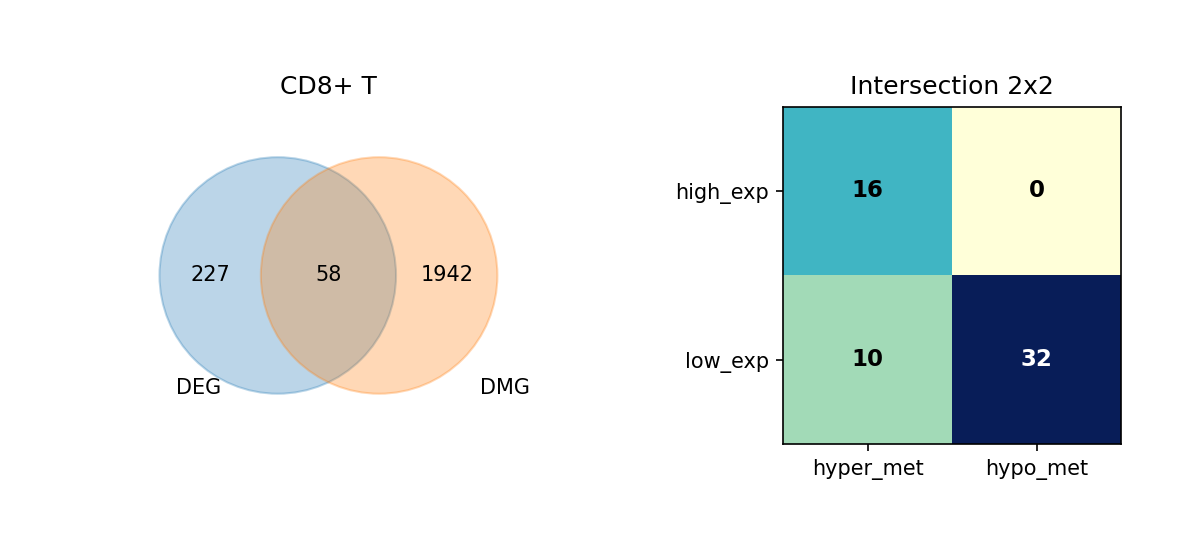
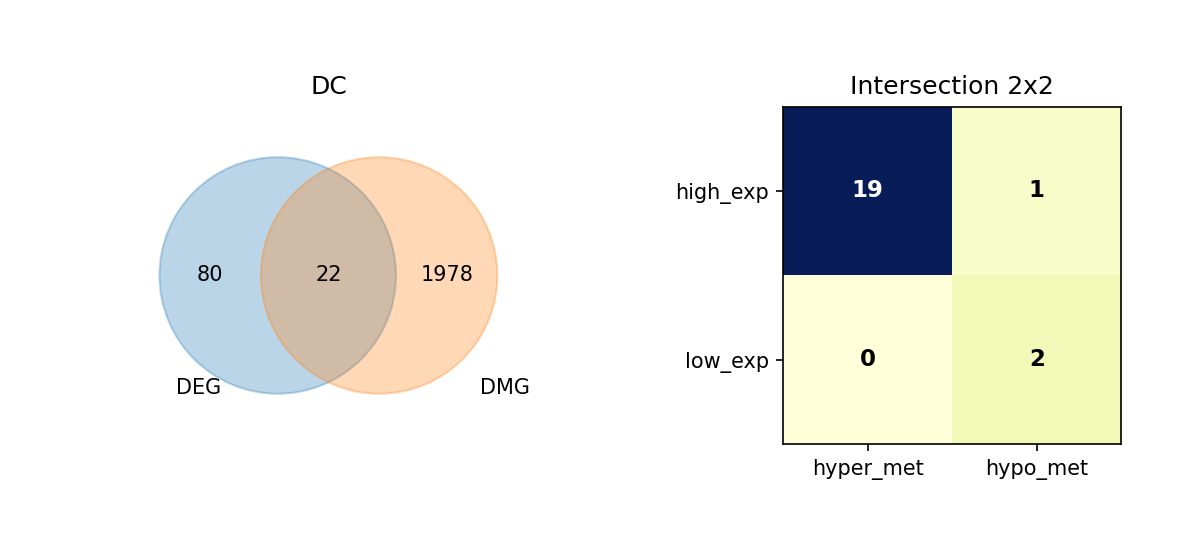
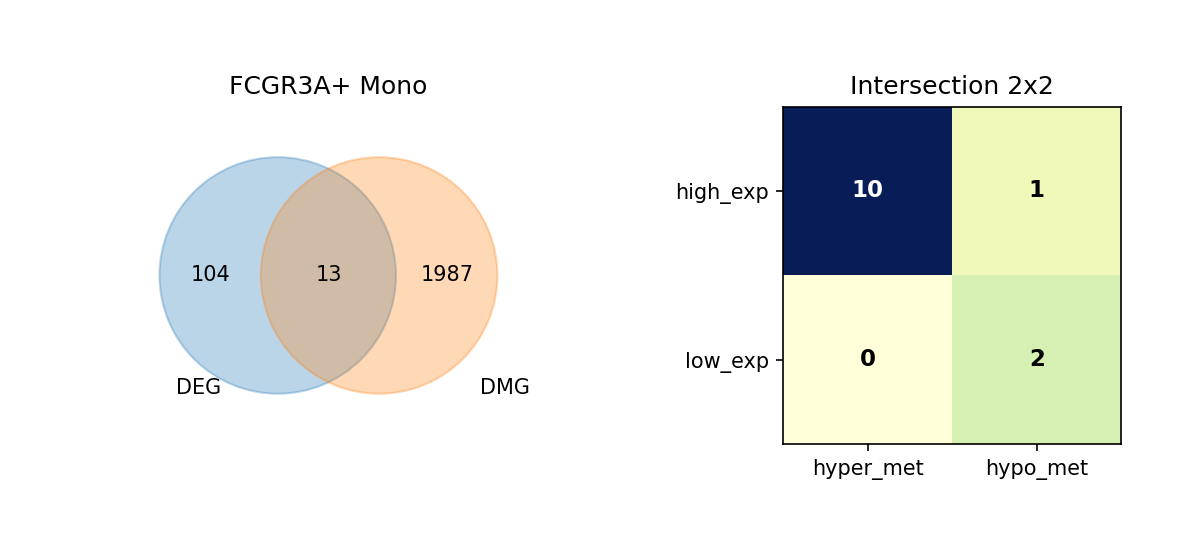
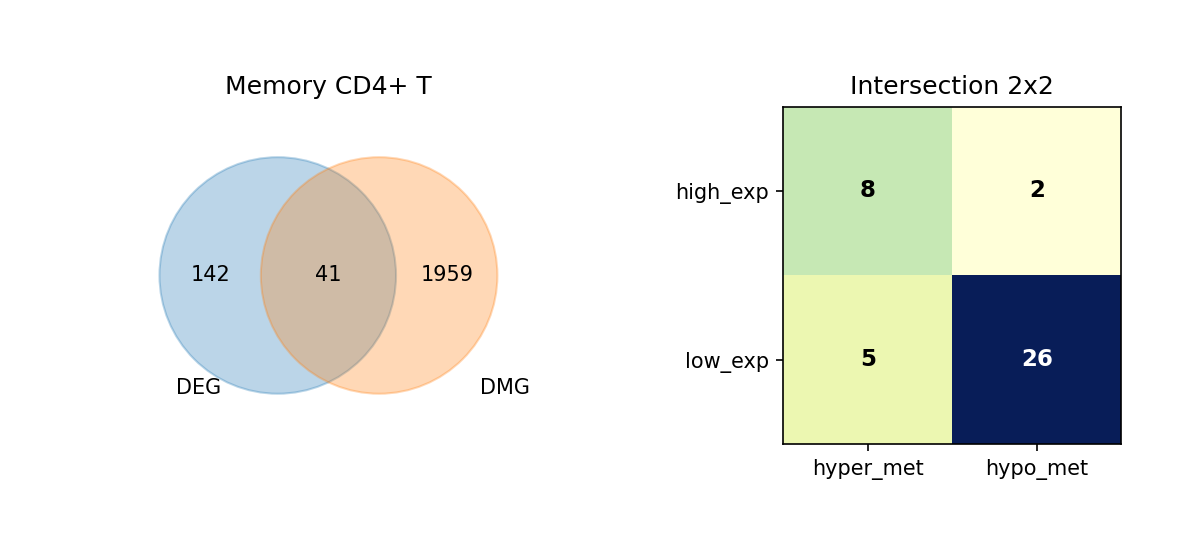
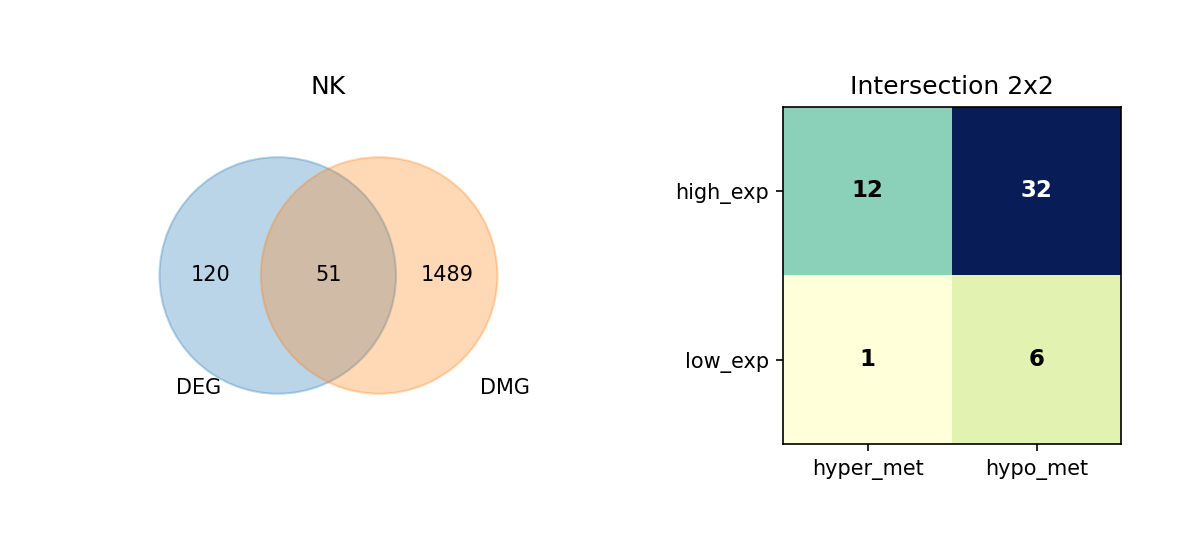
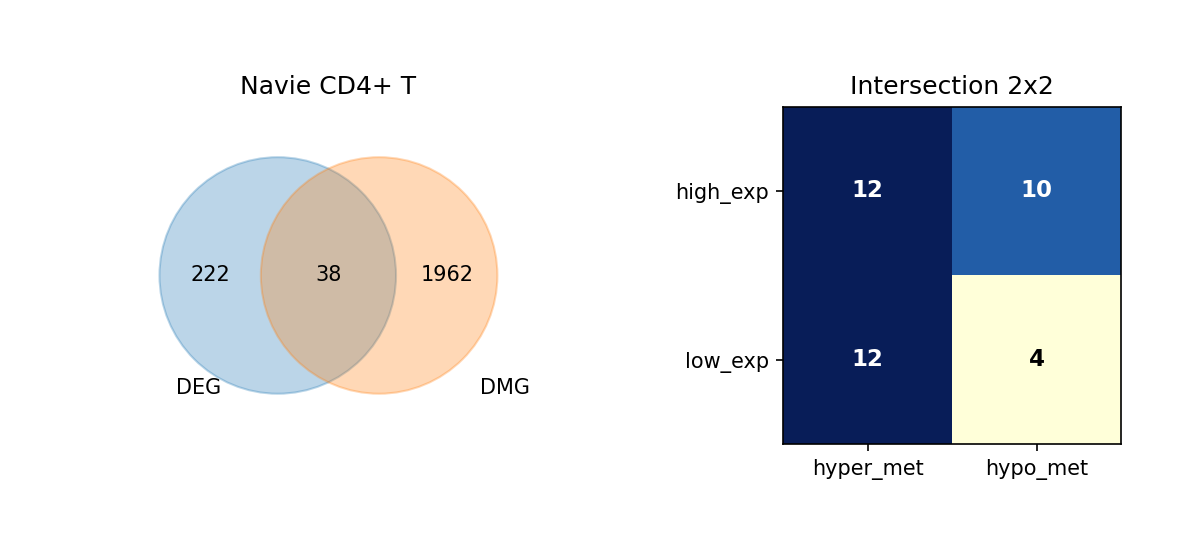
通过维恩图(Venn Diagram),我们展示了每种细胞类型中 DEG 与 DMG 的交集情况,并进一步细分这些重叠基因的表达与甲基化变化方向。
重叠分析的意义:
- 识别关键基因:同时具有差异表达和差异甲基化的基因可能是细胞类型分化和功能的关键调控因子。
- 表观遗传调控:重叠基因可能受到表观遗传修饰的调控,是理解表观遗传-转录调控关系的重要线索。
- 功能关联:重叠基因可能参与细胞类型特异性的生物学过程,值得进一步深入研究。
图注:本图展示每种细胞类型中 DEG 与 DMG 的重叠情况,以及重叠基因的表达与甲基化变化方向。
- 左侧 Venn 图:DEG 与 DMG 的重叠;左侧仅 DEG、右侧仅 DMG、中间为重叠基因数量
- 右侧 2×2 矩阵热图:重叠基因按表达方向与甲基化方向细分
- 行:表达方向(high_exp 高表达、low_exp 低表达)
- 列:甲基化方向(hyper_met 高甲基化、hypo_met 低甲基化)
- 格子数值:该组合下的基因数量;颜色越深(蓝)表示数量越多
HAS_VENN = False
def make_plot_for_cluster(cluster, deg_df, dmg_df, outdir):
"""为每个cluster绘制Venn图和2x2矩阵"""
deg_sub = deg_df[deg_df['cluster'] == cluster].copy()
dmg_sub = dmg_df[dmg_df['met_cluster'] == cluster].copy()
deg_set = set(deg_sub['gene_ids'])
dmg_set = set(dmg_sub['met_gene_ids'])
inter = deg_set & dmg_set
deg_only = len(deg_set - inter)
dmg_only = len(dmg_set - inter)
inter_count = len(inter)
if 'met_status' not in dmg_sub.columns and 'met_fc' in dmg_sub.columns:
dmg_sub['met_status'] = np.where(dmg_sub['met_fc'] >= 1, 'hyper_met', 'hypo_met')
inter_df = pd.merge(deg_sub[['gene_ids','exp_status']],
dmg_sub[['met_gene_ids','met_status']],
left_on='gene_ids', right_on='met_gene_ids', how='inner')
inter_df = inter_df[inter_df['exp_status'] != 'neutral']
def count(exp, met):
return int(((inter_df['exp_status'] == exp) & (inter_df['met_status'] == met)).sum())
counts = {
('high_exp','hyper_met'): count('high_exp','hyper_met'),
('high_exp','hypo_met'): count('high_exp','hypo_met'),
('low_exp','hyper_met'): count('low_exp','hyper_met'),
('low_exp','hypo_met'): count('low_exp','hypo_met')
}
fig, axes = plt.subplots(1, 2, figsize=(8, 3), dpi = 150)
ax = axes[0]
if HAS_VENN:
from matplotlib_venn import venn2
v = venn2(subsets=(deg_only, dmg_only, inter_count), set_labels=('DEG', 'DMG'), ax=ax)
else:
from matplotlib.patches import Circle
ax.set_aspect('equal')
c1 = Circle((0.6, 0.5), 0.35, color='C0', alpha=0.3)
c2 = Circle((0.9, 0.5), 0.35, color='C1', alpha=0.3)
ax.add_patch(c1)
ax.add_patch(c2)
ax.text(0.4, 0.5, str(deg_only), ha='center', va='center')
ax.text(0.75, 0.5, str(inter_count), ha='center', va='center')
ax.text(1.1, 0.5, str(dmg_only), ha='center', va='center')
ax.set_xlim(0, 1.5)
ax.set_ylim(0, 1)
ax.axis('off')
ax.text(0.3, 0.15, 'DEG')
ax.text(1.2, 0.15, 'DMG')
ax.set_title(cluster)
ax2 = axes[1]
matrix = np.array([[counts[('high_exp','hyper_met')], counts[('high_exp','hypo_met')]],
[counts[('low_exp','hyper_met')], counts[('low_exp','hypo_met')]]])
im = ax2.imshow(matrix, cmap='YlGnBu')
thr = matrix.max() / 2 if matrix.max() > 0 else 0
for i in range(2):
for j in range(2):
color = 'white' if matrix[i, j] > thr else 'black'
ax2.text(j, i, matrix[i, j], ha='center', va='center', color=color, fontsize=11, fontweight='bold')
ax2.set_xticks([0, 1])
ax2.set_xticklabels(['hyper_met', 'hypo_met'])
ax2.set_yticks([0, 1])
ax2.set_yticklabels(['high_exp', 'low_exp'])
ax2.set_title('Intersection 2x2')
fig.tight_layout()
plt.show()
return {'deg_only': deg_only, 'dmg_only': dmg_only, 'inter': inter_count, 'counts': counts}
# 为每个cluster绘制Venn图
results = {}
for cluster in clusters:
results[cluster] = make_plot_for_cluster(cluster, deg_table, dmg_table_renamed, "./")
# 生成汇总表格
summary_df = pd.DataFrame.from_dict({k: v['counts'] for k, v in results.items()}, orient='index')
summary_df.columns = [ (f"{c[0]}|{c[1]}" if isinstance(c, tuple) else str(c)) for c in summary_df.columns ]
summary_df.to_csv('summary_counts.csv')
print("Venn图分析完成,结果已保存到 summary_counts.csv")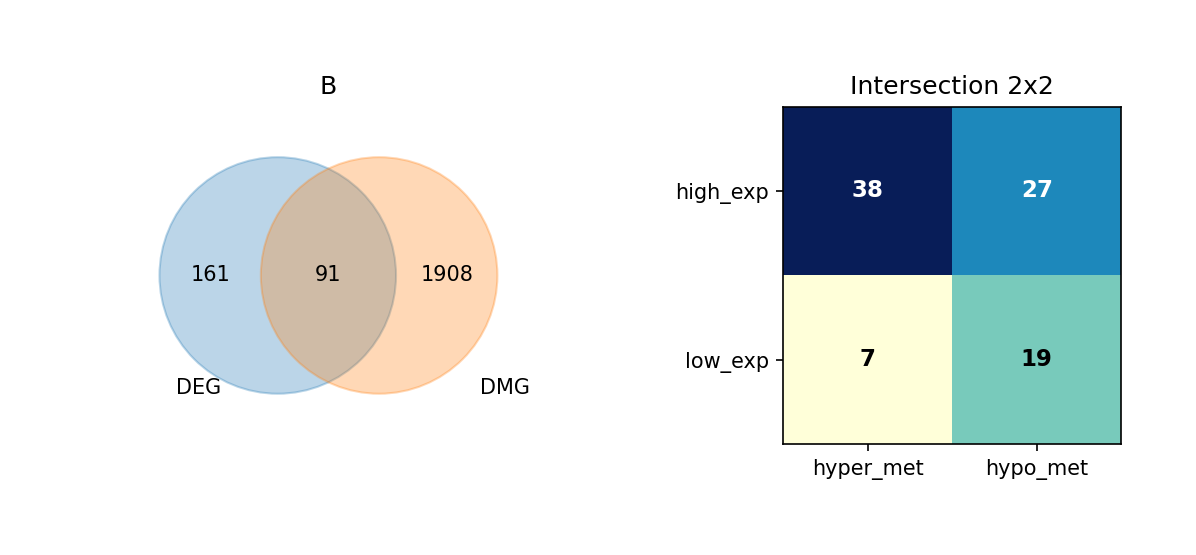
Venn图分析完成,结果已保存到 summary_counts.csv
图注:本图展示所有细胞类型中 DEG 与 DMG 重叠基因的表达/甲基化方向汇总。
- 横轴:表达与甲基化方向的组合(如 high_exp|hyper_met、low_exp|hypo_met 等)
- 纵轴:细胞类型(celltype)
- 热图颜色:表示基因数量,越深(蓝)越多,越浅(黄/白)越少
- 格子数值:该细胞类型在该方向组合下的基因数量;右侧颜色条为数值范围
# 绘制汇总热图
cols_order = ['high_exp|hyper_met','high_exp|hypo_met','low_exp|hyper_met','low_exp|hypo_met']
plot_df = summary_df.reindex(columns=cols_order)
fig, ax = plt.subplots(figsize=(4,4))
im = ax.imshow(plot_df.values, cmap='YlGnBu', aspect='auto')
ax.set_xticks(range(len(cols_order)))
ax.set_xticklabels(cols_order, rotation=45, ha='right')
ax.set_yticks(range(len(plot_df)))
ax.set_yticklabels(plot_df.index)
thr = plot_df.values.max() / 2 if plot_df.values.max() > 0 else 0
for i in range(plot_df.shape[0]):
for j in range(plot_df.shape[1]):
val = int(plot_df.values[i,j])
color = 'white' if val > thr else 'black'
ax.text(j, i, val, ha='center', va='center', color=color, fontsize=10, fontweight='bold')
fig.colorbar(im, ax=ax, fraction=0.046, pad=0.04)
fig.tight_layout()
plt.show()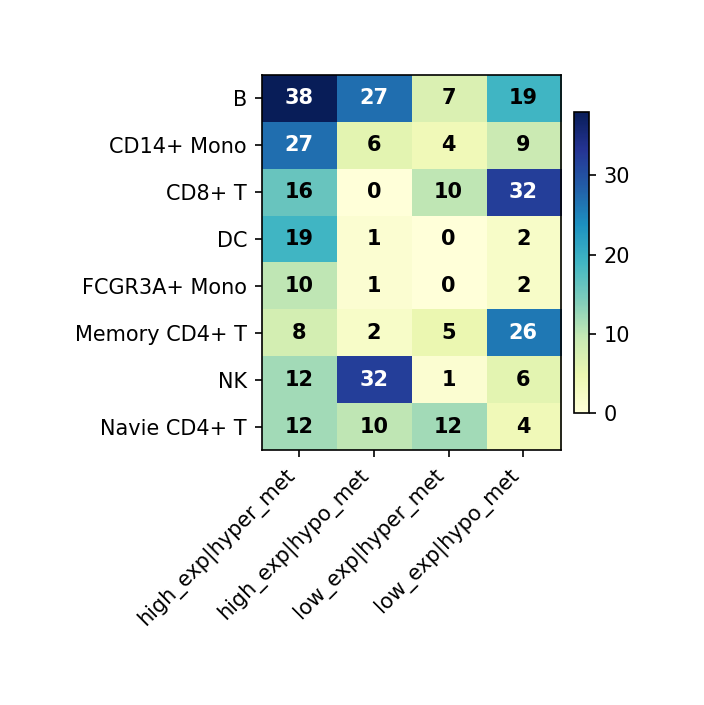
差异基因与差异甲基化基因的 Circos 图展示
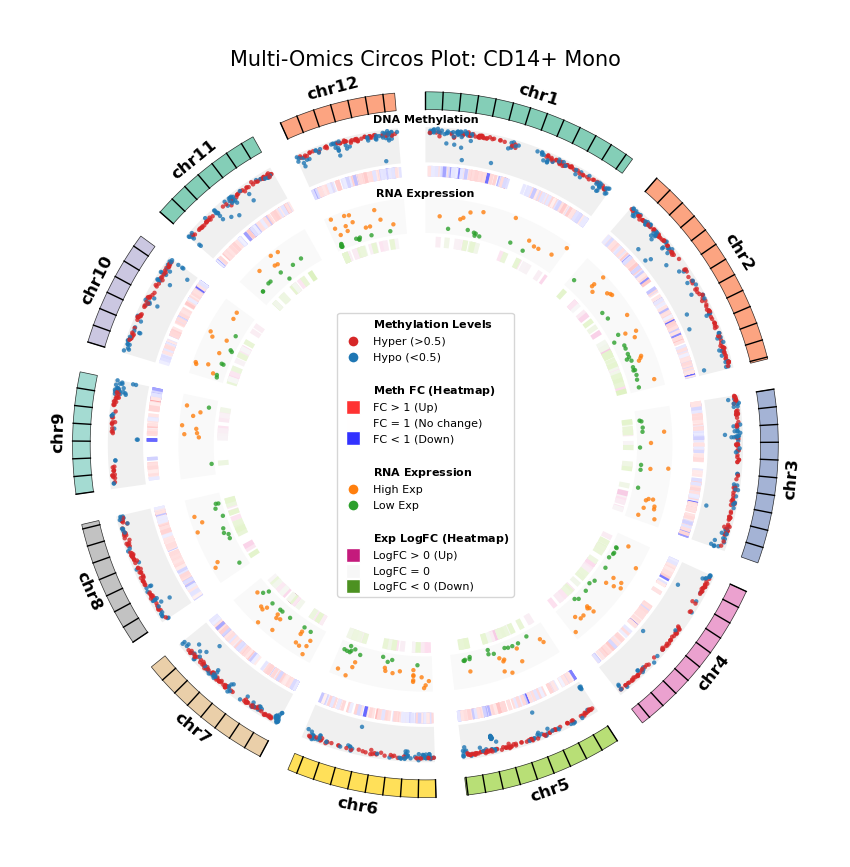
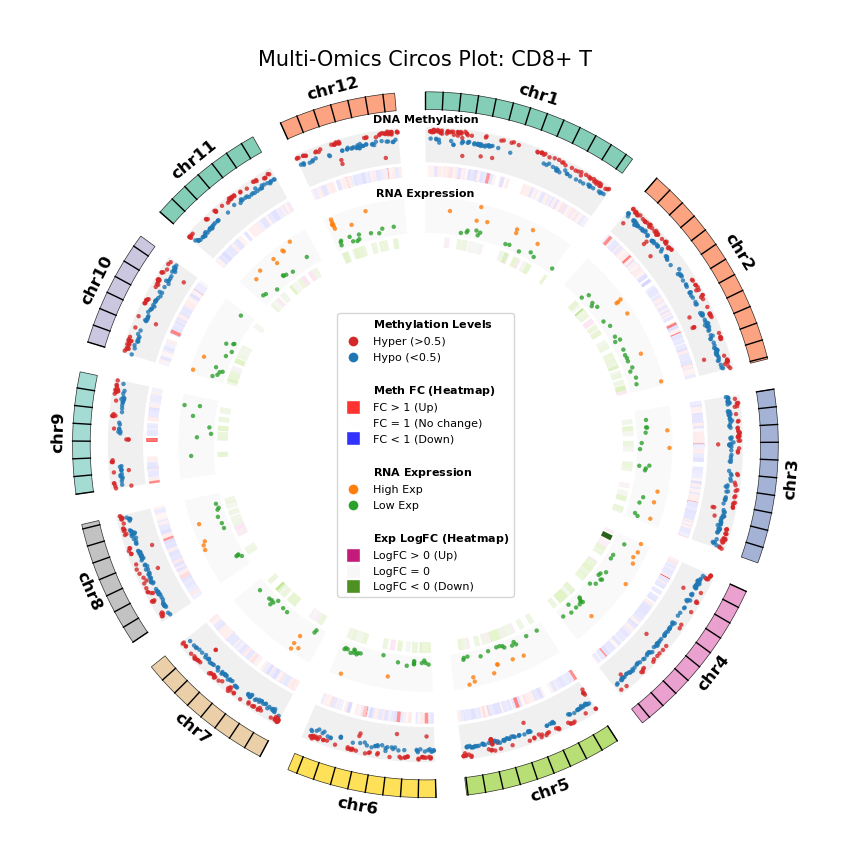
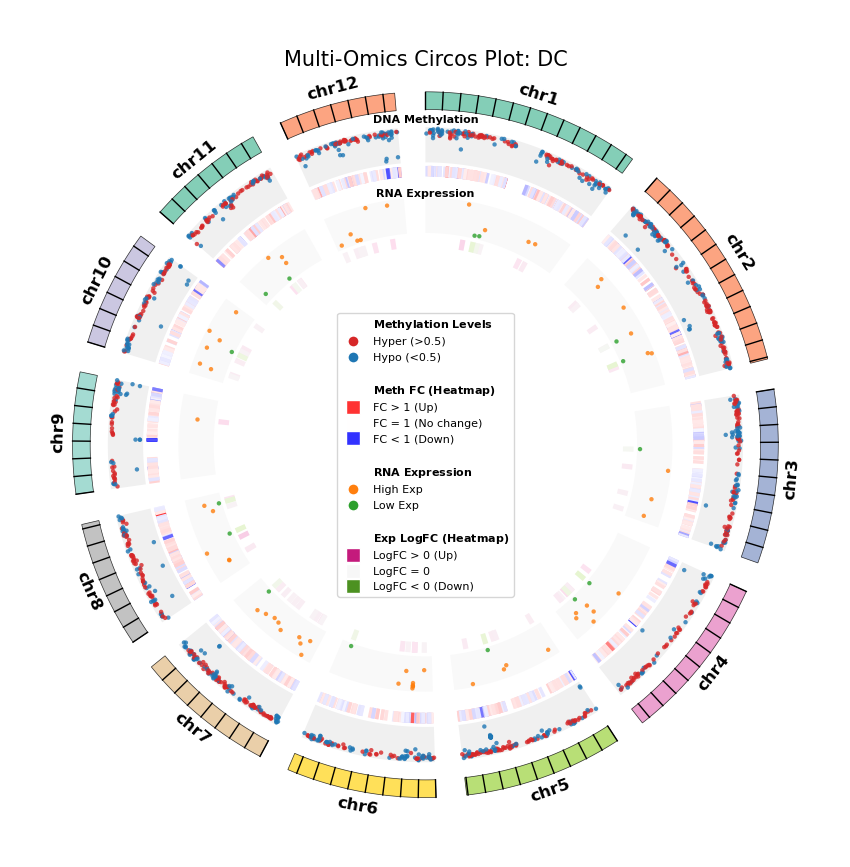
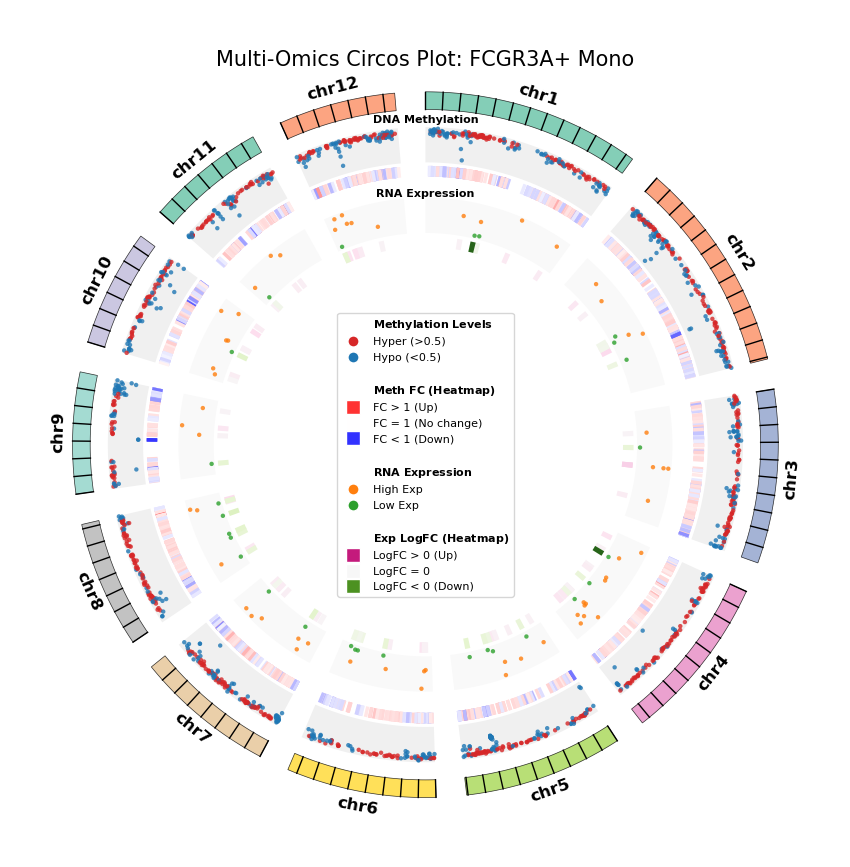
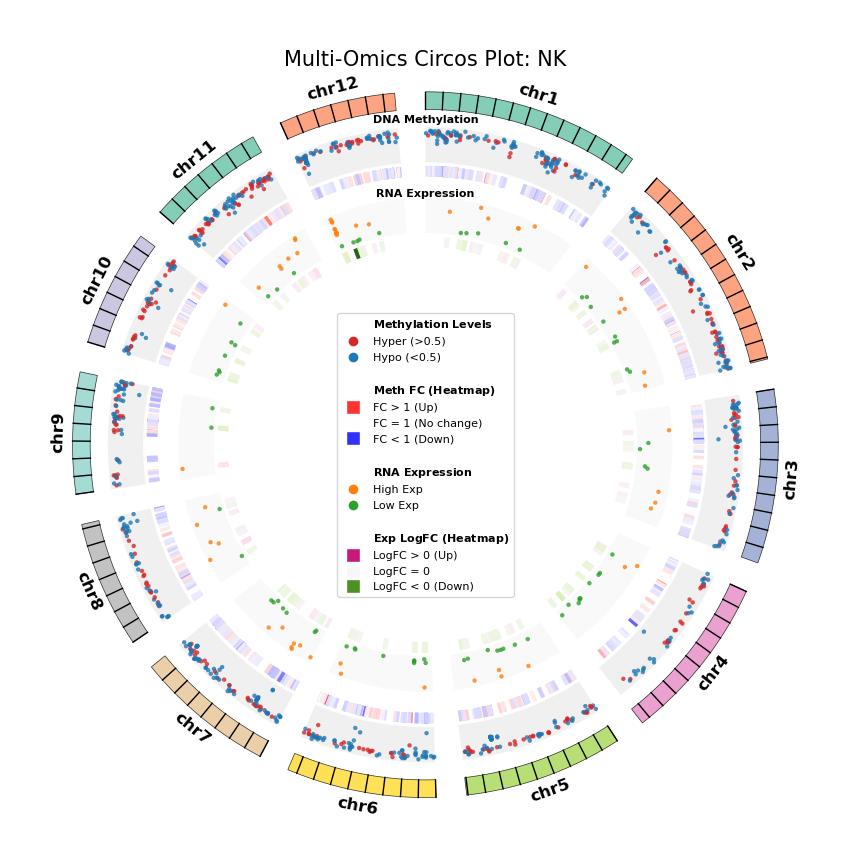
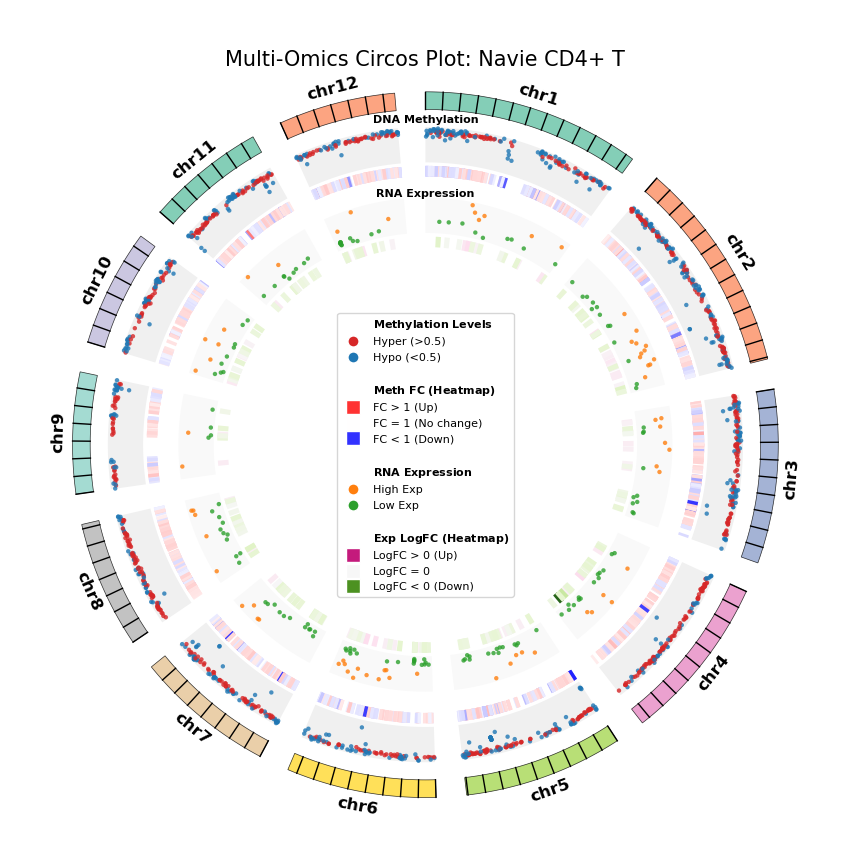
我们利用 Circos 图(Circos Plot)从全基因组尺度展示 DEG 与 DMG 的分布及其对应关系。这种环形可视化方式能够同时展示染色体位置、甲基化变化和基因表达变化,直观地揭示多组学数据的空间关联。
Circos 图的优势:
- 全基因组视角:能够同时展示所有染色体的数据,提供全局视图。
- 空间关联:可以直观地看到 DEG 和 DMG 在基因组上的位置关系,识别共定位模式。
- 多维度信息:在一个图中同时展示位置、甲基化水平和表达水平,便于发现关联。
图注:DNA 甲基化差异基因 (DMG) 与 RNA 差异表达基因 (DEG) 全基因组联合分布图 (Circos Plot)
本图展示了全基因组范围内(选取代表性染色体)DNA 甲基化与基因表达差异的对应关系及空间分布特征。
- 外圈(染色体):展示染色体编号及坐标刻度,不同颜色区分各染色体。
- 中间层(DNA Methylation):
- 散点图:展示 DMG 的平均甲基化水平(0–1)。红色代表高甲基化(Hyper),蓝色代表低甲基化(Hypo)。
- 热图环:展示 DMG 的差异倍数(Fold Change)。红色条带表示甲基化水平上调(FC > 1),蓝色条带表示下调(FC < 1),颜色深浅反映差异幅度。
- 内层(RNA Expression):
- 散点图:展示 DEG 的平均基因表达量。橙色代表高表达(High Exp),绿色代表低表达(Low Exp)。
- 热图环:展示 DEG 的对数差异倍数(Log2FC)。红色/粉色条带表示表达上调(Log2FC > 0),绿色条带表示下调(Log2FC < 0),颜色深浅反映差异幅度。
# ==========================================
# 准备Circos图所需的数据
# ==========================================
# 此步骤为Circos图绘制准备数据,包括:
# 1. 计算每个细胞类型中DMG基因的平均甲基化水平
# 2. 计算每个细胞类型中DEG基因的平均表达水平
# 3. 合并甲基化和表达数据,添加基因组坐标信息
warnings.filterwarnings('ignore')
# ==========================================
# 第一部分:准备甲基化数据(DMG)
# ==========================================
# 过滤甲基化数据,仅保留在combined_gene中存在的细胞
# 确保甲基化数据与基因区域数据一致
adata_met = adata_met[adata_met.obs.index.isin(combined_gene.cell.values)]
# 为geneslop2k_da_true_frac添加细胞类型注释坐标
# geneslop2k_da_true_frac是之前计算的真实甲基化分数(未归一化)
geneslop2k_da_true_frac.coords["celltype"] = ('cell', adata_met.obs["celltype"])
# 构建geneslop2k_ids:将基因ID和基因名用下划线连接
# 格式:gene_id_gene_name(如:ENSG00000128731_HERC2)
dmg_table_renamed['geneslop2k_ids'] = dmg_table_renamed['met_gene_ids'].str.cat(dmg_table_renamed['met_gene_names'], sep = '_')
# 计算每个细胞类型中每个DMG基因的平均甲基化水平
# groupby("celltype").mean(dim='cell'): 按细胞类型分组,计算每个基因的平均甲基化水平
# sel(geneslop2k=...): 仅选择DMG表中的基因
geneslop2k_da_true_frac = geneslop2k_da_true_frac.groupby("celltype").mean(dim='cell').sel(geneslop2k = list(set(dmg_table_renamed['geneslop2k_ids'])))
# 将xarray数据转换为DataFrame,便于后续合并
df = geneslop2k_da_true_frac.compute().to_dataframe().reset_index()
# 将平均甲基化水平合并到DMG表中
# 添加每个细胞类型中每个DMG基因的平均甲基化水平(geneslop2k_da_true_frac列)
dmg_table2 = dmg_table_renamed.merge(df[["celltype",'geneslop2k', 'geneslop2k_da_true_frac']], left_on=['met_cluster', 'geneslop2k_ids'], right_on = ["celltype", 'geneslop2k'], how = 'left')
# ==========================================
# 第二部分:准备表达数据(DEG)
# ==========================================
# 为转录组数据添加geneslop2k_ids,用于后续与甲基化数据匹配
adata_rna.var['geneslop2k_ids'] = adata_rna.var['gene_ids'].str.cat(adata_rna.var.index, sep = '_')
# 获取所有细胞类型
cell_types = adata_rna.obs["celltype"].unique()
# 创建DataFrame用于存储每个细胞类型中每个基因的平均表达量
# 行:细胞类型,列:基因ID
mean_expression = pd.DataFrame(index=cell_types, columns=adata_rna.var['gene_ids'])
# 计算每个细胞类型中每个基因的平均表达量
for ct in cell_types:
# 提取该细胞类型的所有细胞
subset = adata_rna[adata_rna.obs["celltype"] == ct].X
# 处理稀疏矩阵和密集矩阵两种情况
if sparse.issparse(subset):
# 稀疏矩阵:使用.A1将结果转换为1D数组
mean_val = subset.mean(axis=0).A1
else:
# 密集矩阵:直接计算均值
mean_val = subset.mean(axis=0)
# 存入 DataFrame
mean_expression.loc[ct] = mean_val
# 将宽格式转换为长格式,便于后续合并
# melt操作:将行(细胞类型)和列(基因)转换为行,每行包含一个细胞类型-基因对的平均表达量
mean_expression = mean_expression.reset_index().melt(
id_vars='index', # 保留细胞类型列(index)
var_name='gene_ids', # 基因ID列名
value_name='mean_exp') # 平均表达量列名
# 将平均表达量合并到DEG表中
# 添加每个细胞类型中每个DEG基因的平均表达量(mean_exp列)
deg_table2 = deg_table.merge(mean_expression, left_on = ['cluster', 'geneslop2k_gene_ids'], right_on = ['index', 'gene_ids'], how = 'left').drop(['gene_ids_y','index'], axis = 1).rename(columns = {'gene_ids_x':'gene_ids'})for target_cell_type in clusters:
# 过滤数据
plot_meth = dmg_table2[dmg_table2['met_cluster'] == target_cell_type].copy()
plot_exp = deg_table2[deg_table2['cluster'] == target_cell_type].copy()
chrom_sizes = pd.read_table(os.path.join('../../','data', sample_path_config[samples[0]]['top_dir'], 'methylation', sample_path_config[samples[0]]['demo_dir'], samples[0], 'allcools_generate_datasets', f'{samples[0]}.mcds', 'chrom_sizes.txt'), header = None, sep = '\t')
# chrom_sizes 为两列(染色体名、长度),按行构建 sectors,且长度转为 int 供 Circos 使用
def natural_sort_key(chrom_name):
# 去掉 'chr' 前缀(如果有的话),并尝试转为数字
# 比如 "chr1" -> 1, "chr10" -> 10
# "chrX" -> 23, "chrY" -> 24 (给非数字染色体赋予一个很大的数,让它们排在最后)
import re
# 提取名字里的数字部分
match = re.search(r'(\d+)', chrom_name)
if match:
return int(match.group(1))
# 如果没有数字(比如 X, Y, M),手动指定顺序
special_chroms = {'X': 100, 'Y': 101, 'M': 102, 'MT': 102,
'chrX': 100, 'chrY': 101, 'chrM': 102}
return special_chroms.get(chrom_name, 999) # 未知的排到最后
# 2. 读取并排序
sectors_all = {str(row[0]): int(row[1]) for _, row in chrom_sizes.iterrows()}
# 关键修改:使用 key=lambda x: natural_sort_key(x[0]) 进行排序
# sectors = dict(sorted(sectors.items(), key=lambda x: natural_sort_key(x[0])))
# sectors = {str(row[0]): int(row[1]) for _, row in chrom_sizes.iterrows()}
# sectors = dict(sorted(sectors.items()))
#print(f"Sectors defined: {sectors}")
sorted_keys = sorted(sectors_all.keys(), key=natural_sort_key)
top_n = 12
sectors = {k: sectors_all[k] for k in sorted_keys[:top_n]}
circos = Circos(sectors, space=5)
# ==========================================
# 准备颜色映射 (Color Maps)
# ==========================================
# 1. 甲基化 Fold Change 颜色映射 (Center = 1)
# 0 (低) -> 1 (白/灰) -> >1 (高)
min_meth = plot_meth['met_fc'].min()
max_meth = plot_meth['met_fc'].max()
delta_meth = max(abs(1 - min_meth), abs(max_meth - 1))
# 稍微缩一点点范围 (比如 *0.9),让极值颜色更饱和,不用非得等到最大值才红
limit_meth = delta_meth * 0.9
norm_meth_fc = mcolors.TwoSlopeNorm(
vmin=1 - limit_meth,
vcenter=1,
vmax=1 + limit_meth
)
cmap_meth_fc = plt.get_cmap('bwr') # Blue-White-Red
chr_cmap = mcolors.ListedColormap(my_palette)
sector_colors = {name: chr_cmap(i % 10) for i, name in enumerate(sectors.keys())}
# 1. 甲基化 Fold Change 颜色映射 (Center = 1)
# 0 (低) -> 1 (白/灰) -> >1 (高)
# 使用 TwoSlopeNorm 确保 1 对应中间的白色
# norm_meth_fc = mcolors.TwoSlopeNorm(vmin=0, vcenter=1, vmax=2)
# 注意:vmax设为2表示fc=2时达到最红,你可以根据数据调整,比如 plot_meth['met_fc'].max()
# cmap_meth_fc = plt.get_cmap('bwr') # Blue-White-Red
# 2. 表达 Log Fold Change 颜色映射 (Center = 0)
# <0 (低) -> 0 (白/灰) -> >0 (高)
# vmax_exp = max(abs(plot_exp['logfoldchanges'].min()), abs(plot_exp['logfoldchanges'].max()))
# norm_exp_fc = mcolors.TwoSlopeNorm(vmin=-vmax_exp, vcenter=0, vmax=vmax_exp)
# cmap_exp_fc = plt.get_cmap('PiYG_r') # Purple-Green (Green for low, Purple/Red for high), or use 'coolwarm'
min_exp = plot_exp['logfoldchanges'].min()
max_exp = plot_exp['logfoldchanges'].max()
# LogFC 中心是 0
# 同样取最大绝对值,保证 0 是白色
limit_exp = max(abs(min_exp), abs(max_exp)) * 0.9 # 同样 *0.9 增强一点饱和度
norm_exp_fc = mcolors.TwoSlopeNorm(
vmin=-limit_exp,
vcenter=0,
vmax=limit_exp
)
cmap_exp_fc = plt.get_cmap('PiYG_r') # Purple-Green
# ==========================================
# 4. 绘制轨道 (Tracks)
# ==========================================
# --- Track 1: 染色体名称与刻度 ---
# for sector in circos.sectors:
# sector.text(sector.name) # 调整文字位置
# track = sector.add_track((98, 100))
# track.axis(fc='lightgrey')
for sector in circos.sectors:
# 1. 染色体名称 (加粗,稍微向外挪)
# sector.text(sector.name, r=102, size=12, weight='bold', color=sector_colors[sector.name])
sector.text(sector.name, r=102, size=12, weight='bold', color="black")
# 2. 染色体条带 (使用我们分配的彩色)
track = sector.add_track((95, 100))
track.axis(fc=sector_colors[sector.name], alpha=0.8) # 80%不透明度,很有质感
# 3. 添加刻度 (每 20Mb 一个大刻度,带数字)
# interval 单位是 bp,这里设为 20,000,000 (20Mb)
# label_size 控制数字大小,label_orientation 调整方向
interval = 20 * 10**6
limit = int(sector.end - sector.start)
for pos in range(0, limit, int(interval)):
# [0, 0.3] 表示在条带宽度的内侧 30% 处画线 (0是内缘,1是外缘)
track.line([pos, pos], [0, 0.3], color='black', lw=1)
# ==========================================
# region A: 甲基化数据区 (DNA Methylation)
# ==========================================
# 规划:
# r=90-95: 散点图 (Level)
# r=86-89: 热图条带 (Fold Change)
for sector in circos.sectors:
# 获取数据
sub_meth = plot_meth[plot_meth['geneslop2k_chrom'] == sector.name]
# --- Track A1: 甲基化水平散点图 ---
track_scatter = sector.add_track((80, 90))
track_scatter.axis(fc='#f0f0f0', ec='none')
# 添加区域标签 (只在第一个sector添加一次,或者找个固定角度添加)
if sector.name == circos.sectors[0].name:
circos.text('DNA Methylation', r=92, size=8, color='black', weight='bold')
if not sub_meth.empty:
x = sub_meth['geneslop2k_start'].values
y = sub_meth['geneslop2k_da_true_frac'].values
status = sub_meth['met_met_status'].values
colors = np.where(status == 'hyper_met', '#d62728', '#1f77b4')
track_scatter.scatter(x, y, color=colors, s=10, alpha=0.8, vmin=0, vmax=1)
# --- Track A2: 甲基化 FC 热图条带 ---
track_heatmap = sector.add_track((76, 79))
# track_heatmap.axis(fc="none", ec="none") # 热图背景透明
if not sub_meth.empty:
# 遍历画矩形 (模拟热图)
# width 设为 2000 (geneslop2k) 或稍微宽一点以便看清
for _, row in sub_meth.iterrows():
# start = row['geneslop2k_start']
# end = row['geneslop2k_end'] # 或者是 row['geneslop2k_end']
# val = row['met_fc']
# # 获取颜色
# color = cmap_meth_fc(norm_meth_fc(val))
# 画矩形: rect(start, end, ...)
center = (row['geneslop2k_start'] + row['geneslop2k_end']) / 2
width = 5000000 # 1Mb
new_start = center - (width / 2)
new_end = center + (width / 2)
# 2. 关键修复:防止越界 (Clip)
# 必须限制在 [sector.start, sector.end] 范围内
new_start = max(new_start, sector.start)
new_end = min(new_end, sector.end)
# 3. 如果截断后 start >= end,就不画了 (理论上 width 很大不会发生,但为了安全)
if new_start >= new_end:
continue
val = row['met_fc']
color = cmap_meth_fc(norm_meth_fc(val))
track_heatmap.rect(new_start, new_end, color=color, lw=0)
# ==========================================
# region B: 表达数据区 (RNA Expression)
# ==========================================
# 规划:
# r=70-75: 散点图 (Level)
# r=66-69: 热图条带 (LogFC)
for sector in circos.sectors:
sub_exp = plot_exp[plot_exp['geneslop2k_chrom'] == sector.name]
# --- Track B1: 表达水平散点图 ---
track_scatter = sector.add_track((60, 70))
track_scatter.axis(fc='#f9f9f9', ec='none')
# 添加区域标签
if sector.name == circos.sectors[0].name:
circos.text('RNA Expression', r=71, size=8, color='black', weight='bold')
#if not sub_exp.empty:
# x = sub_exp['geneslop2k_start'].values
# y = sub_exp['mean_exp'].values
# status = sub_exp['exp_status'].values
# colors = np.where(status == 'high_exp', '#ff7f0e', '#2ca02c')
# track_scatter.scatter(x, y, color=colors, s=10, alpha=0.8)
if not sub_exp.empty:
x = sub_exp['geneslop2k_start'].values
y = sub_exp['mean_exp'].values
status = sub_exp['exp_status'].values
colors = np.where(status == 'high_exp', '#ff7f0e', '#2ca02c')
# 解决方案1:手动设置合适的 vmin 和 vmax
y_min, y_max = y.min(), y.max()
# 添加一些边距,确保所有点都在范围内
margin = 0.1 * (y_max - y_min) if y_max != y_min else 0.1
vmin = y_min - margin
vmax = y_max + margin
# 修复:添加 vmin 和 vmax 参数
track_scatter.scatter(x, y, color=colors, s=10, alpha=0.8, vmin=vmin, vmax=vmax)
# --- Track B2: 表达 LogFC 热图条带 ---
track_heatmap = sector.add_track((56, 59))
if not sub_exp.empty:
for _, row in sub_exp.iterrows():
# start = row['geneslop2k_start']
# end = row['geneslop2k_end']
# val = row['logfoldchanges']
# # 获取颜色
# color = cmap_exp_fc(norm_exp_fc(val))
# track_heatmap.rect(start, end, color=color, lw=0)
center = (row['geneslop2k_start'] + row['geneslop2k_end']) / 2
width = 10000000 # 5Mb
new_start = center - (width / 2)
new_end = center + (width / 2)
# 2. 关键修复:防止越界 (Clip)
# 必须限制在 [sector.start, sector.end] 范围内
new_start = max(new_start, sector.start)
new_end = min(new_end, sector.end)
if new_start >= new_end:
continue
val = row['logfoldchanges']
color = cmap_exp_fc(norm_exp_fc(val))
track_heatmap.rect(new_start, new_end, color=color, lw=0)
# ==========================================
# 5. 添加图例
# ==========================================
fig = circos.plotfig()
# --- 构建复杂的组合图例 ---
legend_elements = [
# 标题1
Line2D([0], [0], color='w', label=r'$\bf{Methylation\ Levels}$', markersize=0),
Line2D([0], [0], marker='o', color='w', label='Hyper (>0.5)', markerfacecolor='#d62728', markersize=8),
Line2D([0], [0], marker='o', color='w', label='Hypo (<0.5)', markerfacecolor='#1f77b4', markersize=8),
# 标题2 (FC 热图)
Line2D([0], [0], color='w', label=' ', markersize=0), # 空行
Line2D([0], [0], color='w', label=r'$\bf{Meth\ FC\ (Heatmap)}$', markersize=0),
Line2D([0], [0], marker='s', color='w', label='FC > 1 (Up)', markerfacecolor=cmap_meth_fc(0.9), markersize=10),
Line2D([0], [0], marker='s', color='w', label='FC = 1 (No change)', markerfacecolor=cmap_meth_fc(0.5), markersize=10),
Line2D([0], [0], marker='s', color='w', label='FC < 1 (Down)', markerfacecolor=cmap_meth_fc(0.1), markersize=10),
# 标题3
Line2D([0], [0], color='w', label=' ', markersize=0), # 空行
Line2D([0], [0], color='w', label=r'$\bf{RNA\ Expression}$', markersize=0),
Line2D([0], [0], marker='o', color='w', label='High Exp', markerfacecolor='#ff7f0e', markersize=8),
Line2D([0], [0], marker='o', color='w', label='Low Exp', markerfacecolor='#2ca02c', markersize=8),
# 标题4 (LogFC 热图)
Line2D([0], [0], color='w', label=' ', markersize=0), # 空行
Line2D([0], [0], color='w', label=r'$\bf{Exp\ LogFC\ (Heatmap)}$', markersize=0),
Line2D([0], [0], marker='s', color='w', label='LogFC > 0 (Up)', markerfacecolor=cmap_exp_fc(0.9), markersize=10),
Line2D([0], [0], marker='s', color='w', label='LogFC = 0', markerfacecolor=cmap_exp_fc(0.5), markersize=10),
Line2D([0], [0], marker='s', color='w', label='LogFC < 0 (Down)', markerfacecolor=cmap_exp_fc(0.1), markersize=10),
]
# 图例位置
fig.legend(handles=legend_elements, bbox_to_anchor=(0.5, 0.475), loc='center', fontsize=8, borderaxespad=0.)
plt.title(f"Multi-Omics Circos Plot: {target_cell_type}", fontsize=15)
plt.tight_layout()
#plt.savefig(f"figures/circos_{target_cell_type.replace(' ','_')}.pdf", bbox_inches='tight')
#plt.savefig(f"figures/circos_{target_cell_type.replace(' ','_')}.png", bbox_inches='tight')
plt.show()
plt.close()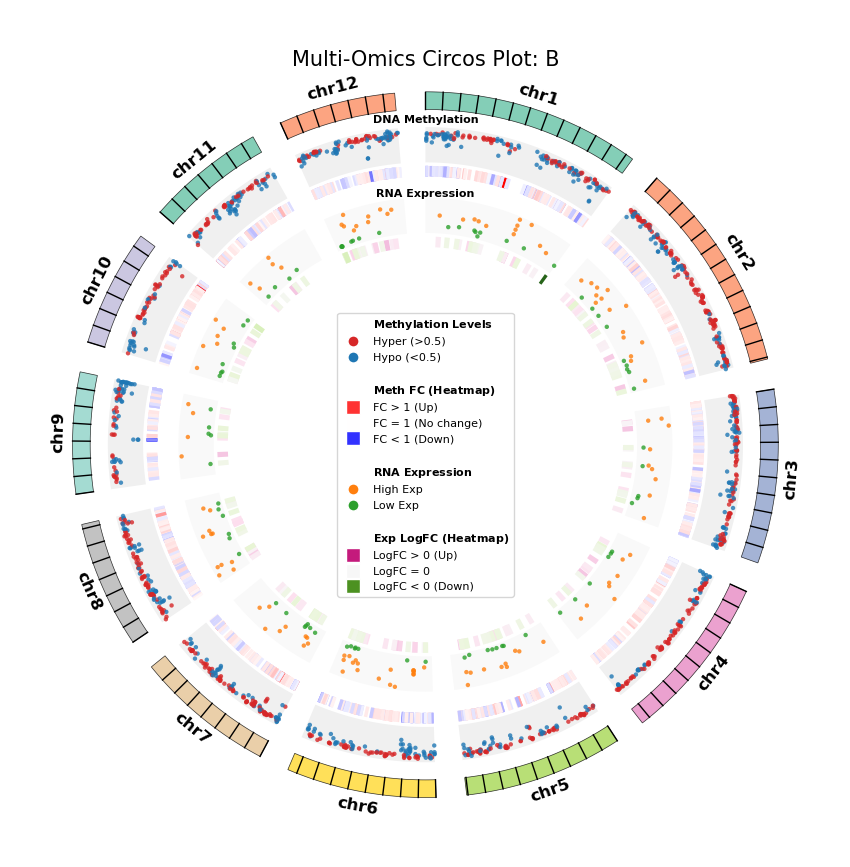

功能富集分析概述
功能富集分析用于识别差异表达基因(DEG)和差异甲基化基因(DMG)在生物学功能、通路等方面的富集情况,帮助理解差异基因的生物学意义。
分析内容:
- GO 富集分析:识别差异基因富集的基因本体(Gene Ontology)功能注释
- GO_Biological_Process (BP):生物学过程。
- GO_Cellular_Component (CC):细胞组成。
- GO_Molecular_Function (MF):分子功能。
- KEGG 富集分析:识别差异基因富集的 KEGG 通路
数据要求:
- DEG.csv:差异表达基因数据,包含列:cluster, names, scores, logfoldchanges, pvals, pvals_adj, pct_nz_group, pct_nz_reference。
- DMG.csv:差异甲基化基因数据,包含列:names, pvals_adj, fc, AUROC, cluster。
💡 Note
本 notebook 利用 Python 包 gseapy 进行基因集功能富集分析。使用 gseapy 可以自动处理基因集、调用 Enrichr 或基于本地 GOA/KEGG 数据库完成注释,并输出标准化结果表格及可视化图表。
使用建议
在进行功能富集分析前,请注意以下事项:
- 前置条件:建议先完成差异分析,获得 DEG 和 DMG 结果后再进行功能富集。
- 网络连接:确保网络连接正常,因为需要访问 Enrichr API。
- 基因数量:如果某个细胞类型的基因数量过少(< 5),将自动跳过该细胞类型的分析。
- 结果保存:富集分析结果会自动保存到指定的输出目录,包括 CSV 表格和可视化图片。
GO 功能富集分析
# 1. 自动识别表格类型
def detect_table_type(df):
"""自动识别表格类型(DEG或DMG)"""
if 'logfoldchanges' in df.columns:
return 'DEG'
elif 'fc' in df.columns:
return 'DMG'
else:
raise ValueError("无法识别表格类型:缺少 logfoldchanges 或 fc 列")
# 2. 预处理基因名(DMG 专用)
def clean_gene_name(gene):
"""清理DMG基因名,提取基因名部分"""
if "_" in gene:
return gene.split("_")[-1]
return gene
# 3. 提取基因列表
def extract_genes(df, table_type, direction='up', logfc_cutoff=0.25, padj_cutoff=0.05):
"""从差异分析结果中提取基因列表"""
df = df.copy()
if table_type == 'DEG':
if direction == 'up':
return df[(df['logfoldchanges'] > logfc_cutoff) & (df['pvals_adj'] < padj_cutoff)]['names'].tolist()
elif direction == 'down':
return df[(df['logfoldchanges'] < -logfc_cutoff) & (df['pvals_adj'] < padj_cutoff)]['names'].tolist()
elif table_type == 'DMG':
df['gene'] = df['names'].apply(clean_gene_name)
if direction == 'up':
return df[(df['fc'] > 1) & (df['pvals_adj'] < padj_cutoff)]['gene'].tolist()
elif direction == 'down':
return df[(df['fc'] < 1) & (df['pvals_adj'] < padj_cutoff)]['gene'].tolist()
raise ValueError("direction 必须是 up 或 down")
# 4. enrichr 富集
def run_enrichr(gene_list, species, analysis_type):
"""使用Enrichr进行功能富集分析"""
sp = species.lower()
if sp in ["human"]:
kegg_lib = "KEGG_2021_Human"
elif sp in ["mouse"]:
kegg_lib = "KEGG_2019_Mouse"
else:
kegg_lib = "KEGG_2016"
if analysis_type == 'GO':
# 分别定义每个GO数据库
go_databases = [
'GO_Biological_Process_2025',
'GO_Cellular_Component_2025',
'GO_Molecular_Function_2025'
]
# 分别调用每个数据库
all_results = []
for db_name in go_databases:
try:
enr = gp.enrichr(
gene_list=gene_list,
gene_sets=[db_name],
organism=species,
outdir=None
)
if enr is None or enr.results is None:
continue
# 处理返回结果
if isinstance(enr.results, pd.DataFrame):
df = enr.results.copy()
elif isinstance(enr.results, dict):
if db_name in enr.results:
df = enr.results[db_name].copy()
else:
df = list(enr.results.values())[0].copy() if enr.results else pd.DataFrame()
else:
continue
if len(df) > 0:
df['Gene_set'] = db_name
all_results.append(df)
except Exception as e:
print(f"警告: {db_name} 分析失败: {e}")
continue
# 合并所有结果
if all_results:
return pd.concat(all_results, ignore_index=True)
else:
return pd.DataFrame()
elif analysis_type == 'KEGG':
# KEGG 只有一个数据库,直接调用
enr = gp.enrichr(
gene_list=gene_list,
gene_sets=[kegg_lib],
organism=species,
outdir=None
)
if enr is None or enr.results is None:
return pd.DataFrame()
# 处理返回结果
if isinstance(enr.results, pd.DataFrame):
return enr.results
elif isinstance(enr.results, dict):
if kegg_lib in enr.results:
return enr.results[kegg_lib]
else:
return list(enr.results.values())[0] if enr.results else pd.DataFrame()
else:
return pd.DataFrame()
else:
return pd.DataFrame()
# 5. 富集入口
def run_enrichment(gene_list, species, analysis_type, method):
"""富集分析入口函数"""
if method != 'enrichr':
raise ValueError("当前脚本仅支持 method='enrichr'")
return run_enrichr(gene_list, species, analysis_type)
# 6. 按细胞类型循环富集 + 自动绘图
def loop_enrichment(df, table_type, species='human', analysis_type='GO', method='enrichr',
direction='up', output_dir="./output", top_term=15,
figsize=(4, 10), fontsize=6, dpi=300, max_term_length=50):
"""按细胞类型循环进行富集分析并自动绘图"""
os.makedirs(output_dir, exist_ok=True)
all_results = []
celltype_col = 'cluster' # DEG 与 DMG 表格均使用 cluster 列
# 设置matplotlib参数
plt.rcParams.update({
'font.size': fontsize,
'axes.titlesize': fontsize,
'axes.labelsize': fontsize-3,
'xtick.labelsize': fontsize - 1,
'ytick.labelsize': fontsize-3,
'legend.fontsize': fontsize - 1,
'figure.dpi': dpi,
'savefig.dpi': dpi,
})
for celltype, sub_df in df.groupby(celltype_col):
gene_list = extract_genes(sub_df, table_type, direction=direction)
if len(gene_list) < 5:
continue
try:
res_df = run_enrichment(gene_list, species, analysis_type, method)
if len(res_df) == 0:
print(f"[WARNING] {celltype} {direction} 没有富集结果,跳过")
continue
res_df['celltype'] = celltype
res_df['direction'] = direction
res_df['analysis_type'] = analysis_type
all_results.append(res_df)
# 自动绘图
safe_name = str(celltype).replace("/", "_").replace(" ", "_")
# 清理Term名称
res_df = res_df.copy()
if 'Term' in res_df.columns:
res_df['Term'] = res_df['Term'].str.split(" \(GO").str[0]
res_df['Term'] = res_df['Term'].str.split(" \(REACTOME").str[0]
res_df['Term'] = res_df['Term'].str.split(" \(KEGG").str[0]
# 处理过长的term名称
def wrap_term(term, max_len=max_term_length):
if len(term) <= max_len:
return term
words = term.split()
lines = []
current_line = ""
for word in words:
test_line = current_line + " " + word if current_line else word
if len(test_line) <= max_len:
current_line = test_line
else:
if current_line:
lines.append(current_line)
current_line = word
if current_line:
lines.append(current_line)
return '\n'.join(lines) if len(lines) > 1 else term[:max_len-3] + '...'
res_df['Term'] = res_df['Term'].apply(wrap_term)
# 按p值排序并取top N
pval_col = 'Adjusted P-value' if 'Adjusted P-value' in res_df.columns else 'P-value'
plot_df = res_df.sort_values(pval_col).head(top_term)
dot_path = os.path.join(output_dir, f"{safe_name}_{direction}_dotplot.png")
bar_path = os.path.join(output_dir, f"{safe_name}_{direction}_barplot.png")
# Dotplot
plt.figure(figsize=figsize, dpi=dpi)
try:
dotplot(plot_df, cmap='viridis_r', size=7, figsize=figsize, top_term=top_term)
fig = plt.gcf()
ax = plt.gca()
fig.set_size_inches(figsize[0], figsize[1])
ax.tick_params(axis='y', labelsize=fontsize)
for label in ax.get_yticklabels():
label.set_fontsize(fontsize)
ax.xaxis.label.set_fontsize(fontsize-3)
ax.tick_params(axis='x', labelsize=fontsize-1)
fig.savefig(dot_path, dpi=dpi, bbox_inches='tight', pad_inches=0.1)
except Exception as e:
print(f"Dotplot绘图失败 {celltype}: {e}")
finally:
plt.close('all')
# Barplot
plt.figure(figsize=figsize, dpi=dpi)
try:
barplot(plot_df, cmap='viridis_r', size=4, color='darkred', figsize=figsize, top_term=top_term)
fig = plt.gcf()
ax = plt.gca()
fig.set_size_inches(figsize[0], figsize[1])
ax.tick_params(axis='y', labelsize=fontsize)
for label in ax.get_yticklabels():
label.set_fontsize(fontsize)
ax.xaxis.label.set_fontsize(fontsize-3)
ax.tick_params(axis='x', labelsize=fontsize-1)
fig.savefig(bar_path, dpi=dpi, bbox_inches='tight', pad_inches=0.1)
except Exception as e:
print(f"Barplot绘图失败 {celltype}: {e}")
finally:
plt.close('all')
print(f"[OK] {celltype} {direction} 图已保存到 {output_dir} (显示 {len(plot_df)} 个term)")
except Exception as e:
print(f"富集失败 {celltype}: {e}")
import traceback
traceback.print_exc()
plt.close('all')
return pd.concat(all_results, ignore_index=True) if all_results else pd.DataFrame()
# 7. 总控函数
def run_full_pipeline(df, species='human', analysis_type='GO', method='enrichr',
direction='both', output_dir="./output",
top_term=15, figsize=(4, 7), fontsize=10, dpi=300):
"""功能富集分析总控函数"""
table_type = detect_table_type(df)
if direction == 'both':
up = loop_enrichment(df, table_type, species, analysis_type, method,
direction='up', output_dir=output_dir,
top_term=top_term, figsize=figsize, fontsize=fontsize, dpi=dpi)
down = loop_enrichment(df, table_type, species, analysis_type, method,
direction='down', output_dir=output_dir,
top_term=top_term, figsize=figsize, fontsize=fontsize, dpi=dpi)
results = []
if not up.empty:
results.append(up)
if not down.empty:
results.append(down)
return pd.concat(results, ignore_index=True) if results else pd.DataFrame()
else:
return loop_enrichment(df, table_type, species, analysis_type, method,
direction=direction, output_dir=output_dir,
top_term=top_term, figsize=figsize, fontsize=fontsize, dpi=dpi)
print("功能富集分析函数定义完成")什么是 GO 富集分析
基因本体(Gene Ontology, GO) 是一个描述基因功能的标准化词汇系统。GO 富集分析是一种常用的功能注释方法,用于识别差异基因集中显著富集的生物学功能。
GO 富集分析的基本原理:
- 通过超几何检验或 Fisher 精确检验,比较差异基因集与背景基因集在某个 GO term 中的分布。
- 计算 p 值来评估差异基因在该功能中是否显著富集。
- 使用多重检验校正(如 FDR)来控制假阳性率。
图注:本图展示差异基因的 GO 功能富集结果(气泡图 Dotplot + 条形图 Barplot)。
- 气泡图:横轴为 Combined Score(综合评分),纵轴为 GO term 名称(按 Adjusted P-value 排序);气泡大小表示基因比例(%Genes in set),颜色表示显著性(log(Adjusted P-value),越深越显著)
- 条形图:横轴为 Adjusted P-value(负对数),纵轴为 GO term 名称
- 图中默认显示 Top 15 个 GO terms(BP/CC/MF),均满足 Adjusted P-value < 0.05
# 读取DEG数据
df_deg = pd.read_csv(deg_file)
DEG_up_GO_dir = os.path.join(output_dir, "DEG", "GO_up_plots")
os.makedirs(DEG_up_GO_dir, exist_ok=True)
output_path = os.path.join(output_dir, "DEG")
os.makedirs(output_path, exist_ok=True)
# 执行GO富集分析(上调基因)
result = run_full_pipeline(
df_deg,
output_dir=DEG_up_GO_dir,
species=species, # 物种:'human'=人类, 'mouse'=小鼠
analysis_type='GO', # 'GO', 'KEGG'
method='enrichr', # 'enrichr'
direction='up' # 'up', 'down', 'both'
)
# 保存结果
DEG_up_GO_file_path = os.path.join(output_path, "GO_up_enrichment.csv")
if not result.empty:
result.to_csv(DEG_up_GO_file_path, index=False)
print(f"DEG上调基因GO富集分析完成,结果已保存到 {DEG_up_GO_file_path}")
else:
print("DEG上调基因GO富集分析未发现显著结果")[OK] CD14+ Mono up 图已保存到 ./enrichment_results/DEG/GO_up_plots (显示 15 个term)
[OK] CD8+ T up 图已保存到 ./enrichment_results/DEG/GO_up_plots (显示 15 个term)
[OK] DC up 图已保存到 ./enrichment_results/DEG/GO_up_plots (显示 15 个term)
[OK] FCGR3A+ Mono up 图已保存到 ./enrichment_results/DEG/GO_up_plots (显示 15 个term)
[OK] Memory CD4+ T up 图已保存到 ./enrichment_results/DEG/GO_up_plots (显示 15 个term)
[OK] NK up 图已保存到 ./enrichment_results/DEG/GO_up_plots (显示 15 个term)
[OK] Navie CD4+ T up 图已保存到 ./enrichment_results/DEG/GO_up_plots (显示 15 个term)
DEG上调基因GO富集分析完成,结果已保存到 ./enrichment_results/DEG/GO_up_enrichment.csv
高表达基因 GO 功能富集分析
分析差异表达基因(DEG)中上调基因(logfoldchanges > 0.25,padj < 0.05)的 GO 功能富集。
# Define image display function (Markdown table version, compatible with VitePress)
def display_go_plots_html_table(output_dir):
"""Read saved dotplot/barplot images from output_dir, display using Markdown table to avoid HTML/Vue conflict"""
from IPython.display import display as ipy_display, Markdown
import base64
import os
import re
import pandas as pd
if not os.path.exists(output_dir):
print(f"Directory does not exist: {output_dir}")
return
# Match filename format: celltype_direction_plot.png
pattern = re.compile(r"^(.+)_(up|down)_(dotplot|barplot)\.png$")
png_files = [f for f in os.listdir(output_dir) if f.endswith(".png")]
if len(png_files) == 0:
print(f"Warning: No PNG image files found in directory {output_dir}")
print(f"Tip: Please ensure enrichment analysis code has run and image files were successfully generated")
return
# Structure: {celltype: {"dotplot": None, "barplot": None}}
plot_dict = {}
for f in png_files:
m = pattern.match(f)
if not m:
continue
celltype = m.group(1)
plot_type = m.group(3)
abs_path = os.path.join(output_dir, f)
if celltype not in plot_dict:
plot_dict[celltype] = {"dotplot": None, "barplot": None}
plot_dict[celltype][plot_type] = abs_path
if len(plot_dict) == 0:
print(f"Warning: Found {len(png_files)} PNG files in directory {output_dir}, but none match expected format")
return
# Generate rows for Markdown table
rows = []
for celltype, plots in plot_dict.items():
dot_html = "Image not found"
bar_html = "Image not found"
if plots["dotplot"] is not None and os.path.exists(plots["dotplot"]):
with open(plots["dotplot"], "rb") as img_file:
img_data = base64.b64encode(img_file.read()).decode()
img_ext = os.path.splitext(plots["dotplot"])[1][1:]
dot_html = f'<img src="/placeholder.svg" style="max-width: 100%; height: auto;" />'
if plots["barplot"] is not None and os.path.exists(plots["barplot"]):
with open(plots["barplot"], "rb") as img_file:
img_data = base64.b64encode(img_file.read()).decode()
img_ext = os.path.splitext(plots["barplot"])[1][1:]
bar_html = f'<img src="/placeholder.svg" style="max-width: 100%; height: auto;" />'
rows.append({
"Cluster": celltype,
"GO_dot": dot_html,
"GO_bar": bar_html
})
rows.sort(key=lambda x: x["Cluster"])
md_table = "| Cluster | GO_dot | GO_bar |\n| :--- | :--- | :--- |\n"
for row in rows:
md_table += f"| {row['Cluster']} | {row['GO_dot']} | {row['GO_bar']} |\n"
ipy_display(Markdown(md_table))图注:本图展示上调基因(logfoldchanges > 0.25,padj < 0.05)的 GO 富集结果;图元含义见上文「GO 功能富集分析可视化图」图注。
# 执行GO富集分析(下调基因)
df_deg = pd.read_csv(deg_file)
DEG_down_GO_dir = os.path.join(output_dir, "DEG", "GO_down_plots")
os.makedirs(DEG_down_GO_dir, exist_ok=True)
result = run_full_pipeline(
df_deg,
output_dir=DEG_down_GO_dir,
species=species,
analysis_type='GO',
method='enrichr',
direction='down'
)
# 保存结果
DEG_down_GO_file_path = os.path.join(output_path, "GO_down_enrichment.csv")
if not result.empty:
result.to_csv(DEG_down_GO_file_path, index=False)
print(f"DEG下调基因GO富集分析完成,结果已保存到 {DEG_down_GO_file_path}")
else:
print("DEG下调基因GO富集分析未发现显著结果")[OK] CD14+ Mono down 图已保存到 ./enrichment_results/DEG/GO_down_plots (显示 15 个term)
[OK] CD8+ T down 图已保存到 ./enrichment_results/DEG/GO_down_plots (显示 15 个term)
[OK] DC down 图已保存到 ./enrichment_results/DEG/GO_down_plots (显示 15 个term)
[OK] FCGR3A+ Mono down 图已保存到 ./enrichment_results/DEG/GO_down_plots (显示 15 个term)
[OK] Memory CD4+ T down 图已保存到 ./enrichment_results/DEG/GO_down_plots (显示 15 个term)
[OK] NK down 图已保存到 ./enrichment_results/DEG/GO_down_plots (显示 15 个term)
[OK] Navie CD4+ T down 图已保存到 ./enrichment_results/DEG/GO_down_plots (显示 15 个term)
DEG下调基因GO富集分析完成,结果已保存到 ./enrichment_results/DEG/GO_down_enrichment.csv
低表达基因 GO 功能富集分析
分析差异表达基因(DEG)中下调基因(logfoldchanges < -0.25,padj < 0.05)的 GO 功能富集。
# 展示DEG下调基因GO富集结果
if os.path.exists(DEG_down_GO_file_path):
display_go_plots_html_table(DEG_down_GO_dir)图注:本图展示下调基因(logfoldchanges < -0.25,padj < 0.05)的 GO 富集结果;图元含义见上文「GO 功能富集分析可视化图」图注。
# 读取DMG数据
df_dmg = pd.read_csv(dmg_file)
DMG_up_GO_dir = os.path.join(output_dir, "DMG", "GO_up_plots")
os.makedirs(DMG_up_GO_dir, exist_ok=True)
output_path_dmg = os.path.join(output_dir, "DMG")
os.makedirs(output_path_dmg, exist_ok=True)
# 执行GO富集分析(高甲基化基因)
result = run_full_pipeline(
df_dmg,
output_dir=DMG_up_GO_dir,
species=species,
analysis_type='GO',
method='enrichr',
direction='up'
)
# 保存结果
DMG_up_GO_file_path = os.path.join(output_path_dmg, "GO_up_enrichment.csv")
if not result.empty:
result.to_csv(DMG_up_GO_file_path, index=False)
print(f"DMG高甲基化基因GO富集分析完成,结果已保存到 {DMG_up_GO_file_path}")
else:
print("DMG高甲基化基因GO富集分析未发现显著结果")[OK] CD14+ Mono up 图已保存到 ./enrichment_results/DMG/GO_up_plots (显示 15 个term)
[OK] CD8+ T up 图已保存到 ./enrichment_results/DMG/GO_up_plots (显示 15 个term)
[OK] DC up 图已保存到 ./enrichment_results/DMG/GO_up_plots (显示 15 个term)
[OK] FCGR3A+ Mono up 图已保存到 ./enrichment_results/DMG/GO_up_plots (显示 15 个term)
[OK] Memory CD4+ T up 图已保存到 ./enrichment_results/DMG/GO_up_plots (显示 15 个term)
Dotplot绘图失败 NK: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 NK: Warning: No enrich terms when cutoff = 0.05
[OK] NK up 图已保存到 ./enrichment_results/DMG/GO_up_plots (显示 15 个term)
[OK] Navie CD4+ T up 图已保存到 ./enrichment_results/DMG/GO_up_plots (显示 15 个term)
DMG高甲基化基因GO富集分析完成,结果已保存到 ./enrichment_results/DMG/GO_up_enrichment.csv
高甲基化基因 GO 功能富集分析
分析差异甲基化基因(DMG)中高甲基化基因(fc > 1,padj < 0.05)的 GO 功能富集。
💡 Note
脚本会自动处理 DMG 数据中的基因名格式(如ENSG00000128731_HERC2),提取基因名部分(HERC2)用于分析。
# 展示DMG高甲基化基因GO富集结果
if os.path.exists(DMG_up_GO_file_path):
display_go_plots_html_table(DMG_up_GO_dir)图注:本图展示高甲基化基因(fc > 1,padj < 0.05)的 GO 富集结果;图元含义见上文「GO 功能富集分析可视化图」图注。
# 执行GO富集分析(低甲基化基因)
df_dmg = pd.read_csv(dmg_file)
DMG_down_GO_dir = os.path.join(output_dir, "DMG", "GO_down_plots")
os.makedirs(DMG_down_GO_dir, exist_ok=True)
result = run_full_pipeline(
df_dmg,
output_dir=DMG_down_GO_dir,
species=species,
analysis_type='GO',
method='enrichr',
direction='down'
)
# 保存结果
DMG_down_GO_file_path = os.path.join(output_path_dmg, "GO_down_enrichment.csv")
if not result.empty:
result.to_csv(DMG_down_GO_file_path, index=False)
print(f"DMG低甲基化基因GO富集分析完成,结果已保存到 {DMG_down_GO_file_path}")
else:
print("DMG低甲基化基因GO富集分析未发现显著结果")Barplot绘图失败 B: Warning: No enrich terms when cutoff = 0.05
[OK] B down 图已保存到 ./enrichment_results/DMG/GO_down_plots (显示 15 个term)
Dotplot绘图失败 CD14+ Mono: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 CD14+ Mono: Warning: No enrich terms when cutoff = 0.05
[OK] CD14+ Mono down 图已保存到 ./enrichment_results/DMG/GO_down_plots (显示 15 个term)
[OK] CD8+ T down 图已保存到 ./enrichment_results/DMG/GO_down_plots (显示 15 个term)
[OK] DC down 图已保存到 ./enrichment_results/DMG/GO_down_plots (显示 15 个term)
Dotplot绘图失败 FCGR3A+ Mono: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 FCGR3A+ Mono: Warning: No enrich terms when cutoff = 0.05
[OK] FCGR3A+ Mono down 图已保存到 ./enrichment_results/DMG/GO_down_plots (显示 15 个term)
[OK] Memory CD4+ T down 图已保存到 ./enrichment_results/DMG/GO_down_plots (显示 15 个term)
Dotplot绘图失败 NK: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 NK: Warning: No enrich terms when cutoff = 0.05
[OK] NK down 图已保存到 ./enrichment_results/DMG/GO_down_plots (显示 15 个term)
[OK] Navie CD4+ T down 图已保存到 ./enrichment_results/DMG/GO_down_plots (显示 15 个term)
DMG低甲基化基因GO富集分析完成,结果已保存到 ./enrichment_results/DMG/GO_down_enrichment.csv
低甲基化基因 GO 功能富集分析
分析差异甲基化基因(DMG)中低甲基化基因(fc < 1,padj < 0.05)的 GO 功能富集。
# 展示DMG低甲基化基因GO富集结果
if os.path.exists(DMG_down_GO_file_path):
display_go_plots_html_table(DMG_down_GO_dir)图注:本图展示低甲基化基因(fc < 1,padj < 0.05)的 GO 富集结果;图元含义见上文「GO 功能富集分析可视化图」图注。
KEGG 功能富集分析
什么是 KEGG 功能富集分析
KEGG(Kyoto Encyclopedia of Genes and Genomes) 是整合了基因组、化学和系统功能信息的综合性数据库。KEGG 富集分析用于识别差异基因集中显著富集的生物学通路。
KEGG 数据库的主要内容:
- KEGG PATHWAY:生物通路图谱,包括代谢通路、信号转导通路、疾病相关通路等
- KEGG GENES:基因和蛋白质的功能注释
- KEGG COMPOUND:化合物数据库
- KEGG DISEASE:疾病相关通路
KEGG 富集分析的基本原理:
- 与 GO 富集分析类似,通过统计检验(如超几何检验)比较差异基因集与背景基因集在某个 KEGG 通路中的分布。
- 识别差异基因显著富集的生物学通路。
- 有助于理解差异基因参与的生物学过程和通路调控机制。
# 执行KEGG富集分析(上调基因)
df_deg = pd.read_csv(deg_file)
DEG_up_KEGG_dir = os.path.join(output_dir, "DEG", "KEGG_up_plots")
os.makedirs(DEG_up_KEGG_dir, exist_ok=True)
result = run_full_pipeline(
df_deg,
output_dir=DEG_up_KEGG_dir,
species=species,
analysis_type='KEGG',
method='enrichr',
direction='up'
)
# 保存结果
DEG_up_KEGG_file_path = os.path.join(output_path, "KEGG_up_enrichment.csv")
if not result.empty:
result.to_csv(DEG_up_KEGG_file_path, index=False)
print(f"DEG上调基因KEGG富集分析完成,结果已保存到 {DEG_up_KEGG_file_path}")
else:
print("DEG上调基因KEGG富集分析未发现显著结果")[OK] CD14+ Mono up 图已保存到 ./enrichment_results/DEG/KEGG_up_plots (显示 15 个term)
[OK] CD8+ T up 图已保存到 ./enrichment_results/DEG/KEGG_up_plots (显示 15 个term)
[OK] DC up 图已保存到 ./enrichment_results/DEG/KEGG_up_plots (显示 15 个term)
[OK] FCGR3A+ Mono up 图已保存到 ./enrichment_results/DEG/KEGG_up_plots (显示 15 个term)
[OK] Memory CD4+ T up 图已保存到 ./enrichment_results/DEG/KEGG_up_plots (显示 15 个term)
[OK] NK up 图已保存到 ./enrichment_results/DEG/KEGG_up_plots (显示 15 个term)
[OK] Navie CD4+ T up 图已保存到 ./enrichment_results/DEG/KEGG_up_plots (显示 15 个term)
DEG上调基因KEGG富集分析完成,结果已保存到 ./enrichment_results/DEG/KEGG_up_enrichment.csv
图注:本图展示差异基因的 KEGG 通路富集结果(气泡图 Dotplot + 条形图 Barplot)。
- 气泡图:横轴为 Combined Score,纵轴为 KEGG 通路名称(按 Adjusted P-value 排序);气泡大小表示基因比例(%Genes in set),颜色表示显著性(log(Adjusted P-value),越深越显著)
- 条形图:横轴为 Adjusted P-value(负对数),纵轴为 KEGG 通路名称
- 图中默认显示 Top 15 个通路,均满足 Adjusted P-value < 0.05;通路 ID(如 hsa04010)可在 KEGG 官网查询详情
# 展示DEG上调基因KEGG富集结果
if os.path.exists(DEG_up_KEGG_file_path):
display_go_plots_html_table(DEG_up_KEGG_dir)高表达基因 KEGG 功能富集分析
分析差异表达基因(DEG)中上调基因(logfoldchanges > 0.25,padj < 0.05)的 KEGG 通路富集。
# 执行KEGG富集分析(下调基因)
df_deg = pd.read_csv(deg_file)
DEG_down_KEGG_dir = os.path.join(output_dir, "DEG", "KEGG_down_plots")
os.makedirs(DEG_down_KEGG_dir, exist_ok=True)
result = run_full_pipeline(
df_deg,
output_dir=DEG_down_KEGG_dir,
species=species,
analysis_type='KEGG',
method='enrichr',
direction='down'
)
# 保存结果
DEG_down_KEGG_file_path = os.path.join(output_path, "KEGG_down_enrichment.csv")
if not result.empty:
result.to_csv(DEG_down_KEGG_file_path, index=False)
print(f"DEG下调基因KEGG富集分析完成,结果已保存到 {DEG_down_KEGG_file_path}")
else:
print("DEG下调基因KEGG富集分析未发现显著结果")[OK] CD14+ Mono down 图已保存到 ./enrichment_results/DEG/KEGG_down_plots (显示 15 个term)
[OK] CD8+ T down 图已保存到 ./enrichment_results/DEG/KEGG_down_plots (显示 15 个term)
[OK] DC down 图已保存到 ./enrichment_results/DEG/KEGG_down_plots (显示 15 个term)
[OK] FCGR3A+ Mono down 图已保存到 ./enrichment_results/DEG/KEGG_down_plots (显示 15 个term)
[OK] Memory CD4+ T down 图已保存到 ./enrichment_results/DEG/KEGG_down_plots (显示 15 个term)
[OK] NK down 图已保存到 ./enrichment_results/DEG/KEGG_down_plots (显示 15 个term)
[OK] Navie CD4+ T down 图已保存到 ./enrichment_results/DEG/KEGG_down_plots (显示 15 个term)
DEG下调基因KEGG富集分析完成,结果已保存到 ./enrichment_results/DEG/KEGG_down_enrichment.csv
图注:本图展示上调基因(logfoldchanges > 0.25,padj < 0.05)的 KEGG 通路富集结果;图元含义见上文「KEGG 功能富集分析可视化图」图注。
# 展示DEG下调基因KEGG富集结果
if os.path.exists(DEG_down_KEGG_file_path):
display_go_plots_html_table(DEG_down_KEGG_dir)低表达基因 KEGG 功能富集分析
分析差异表达基因(DEG)中下调基因(logfoldchanges < -0.25,padj < 0.05)的 KEGG 通路富集。
# 执行KEGG富集分析(高甲基化基因)
df_dmg = pd.read_csv(dmg_file)
DMG_up_KEGG_dir = os.path.join(output_dir, "DMG", "KEGG_up_plots")
os.makedirs(DMG_up_KEGG_dir, exist_ok=True)
result = run_full_pipeline(
df_dmg,
output_dir=DMG_up_KEGG_dir,
species=species,
analysis_type='KEGG',
method='enrichr',
direction='up'
)
# 保存结果
DMG_up_KEGG_file_path = os.path.join(output_path_dmg, "KEGG_up_enrichment.csv")
if not result.empty:
result.to_csv(DMG_up_KEGG_file_path, index=False)
print(f"DMG高甲基化基因KEGG富集分析完成,结果已保存到 {DMG_up_KEGG_file_path}")
else:
print("DMG高甲基化基因KEGG富集分析未发现显著结果")Barplot绘图失败 B: Warning: No enrich terms when cutoff = 0.05
[OK] B up 图已保存到 ./enrichment_results/DMG/KEGG_up_plots (显示 15 个term)
[OK] CD14+ Mono up 图已保存到 ./enrichment_results/DMG/KEGG_up_plots (显示 15 个term)
Dotplot绘图失败 CD8+ T: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 CD8+ T: Warning: No enrich terms when cutoff = 0.05
[OK] CD8+ T up 图已保存到 ./enrichment_results/DMG/KEGG_up_plots (显示 15 个term)
Dotplot绘图失败 DC: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 DC: Warning: No enrich terms when cutoff = 0.05
[OK] DC up 图已保存到 ./enrichment_results/DMG/KEGG_up_plots (显示 15 个term)
Dotplot绘图失败 FCGR3A+ Mono: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 FCGR3A+ Mono: Warning: No enrich terms when cutoff = 0.05
[OK] FCGR3A+ Mono up 图已保存到 ./enrichment_results/DMG/KEGG_up_plots (显示 15 个term)
Dotplot绘图失败 Memory CD4+ T: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 Memory CD4+ T: Warning: No enrich terms when cutoff = 0.05
[OK] Memory CD4+ T up 图已保存到 ./enrichment_results/DMG/KEGG_up_plots (显示 15 个term)
Dotplot绘图失败 NK: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 NK: Warning: No enrich terms when cutoff = 0.05
[OK] NK up 图已保存到 ./enrichment_results/DMG/KEGG_up_plots (显示 15 个term)
Dotplot绘图失败 Navie CD4+ T: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 Navie CD4+ T: Warning: No enrich terms when cutoff = 0.05
[OK] Navie CD4+ T up 图已保存到 ./enrichment_results/DMG/KEGG_up_plots (显示 15 个term)
DMG高甲基化基因KEGG富集分析完成,结果已保存到 ./enrichment_results/DMG/KEGG_up_enrichment.csv
图注:本图展示下调基因(logfoldchanges < -0.25,padj < 0.05)的 KEGG 通路富集结果;图元含义见上文「KEGG 功能富集分析可视化图」图注。
# 展示DMG高甲基化基因KEGG富集结果
if os.path.exists(DMG_up_KEGG_file_path):
display_go_plots_html_table(DMG_up_KEGG_dir)高甲基化基因 KEGG 功能富集分析
分析差异甲基化基因(DMG)中高甲基化基因(fc > 1,padj < 0.05)的 KEGG 通路富集。
# 执行KEGG富集分析(低甲基化基因)
df_dmg = pd.read_csv(dmg_file)
DMG_down_KEGG_dir = os.path.join(output_dir, "DMG", "KEGG_down_plots")
os.makedirs(DMG_down_KEGG_dir, exist_ok=True)
result = run_full_pipeline(
df_dmg,
output_dir=DMG_down_KEGG_dir,
species=species,
analysis_type='KEGG',
method='enrichr',
direction='down'
)
# 保存结果
DMG_down_KEGG_file_path = os.path.join(output_path_dmg, "KEGG_down_enrichment.csv")
if not result.empty:
result.to_csv(DMG_down_KEGG_file_path, index=False)
print(f"DMG低甲基化基因KEGG富集分析完成,结果已保存到 {DMG_down_KEGG_file_path}")
else:
print("DMG低甲基化基因KEGG富集分析未发现显著结果")Dotplot绘图失败 CD14+ Mono: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 CD14+ Mono: Warning: No enrich terms when cutoff = 0.05
[OK] CD14+ Mono down 图已保存到 ./enrichment_results/DMG/KEGG_down_plots (显示 15 个term)
Dotplot绘图失败 CD8+ T: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 CD8+ T: Warning: No enrich terms when cutoff = 0.05
[OK] CD8+ T down 图已保存到 ./enrichment_results/DMG/KEGG_down_plots (显示 15 个term)
Dotplot绘图失败 DC: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 DC: Warning: No enrich terms when cutoff = 0.05
[OK] DC down 图已保存到 ./enrichment_results/DMG/KEGG_down_plots (显示 15 个term)
Dotplot绘图失败 FCGR3A+ Mono: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 FCGR3A+ Mono: Warning: No enrich terms when cutoff = 0.05
[OK] FCGR3A+ Mono down 图已保存到 ./enrichment_results/DMG/KEGG_down_plots (显示 15 个term)
Dotplot绘图失败 Memory CD4+ T: Warning: No enrich terms when cutoff = 0.05
Barplot绘图失败 Memory CD4+ T: Warning: No enrich terms when cutoff = 0.05
[OK] Memory CD4+ T down 图已保存到 ./enrichment_results/DMG/KEGG_down_plots (显示 15 个term)
[OK] NK down 图已保存到 ./enrichment_results/DMG/KEGG_down_plots (显示 15 个term)
[OK] Navie CD4+ T down 图已保存到 ./enrichment_results/DMG/KEGG_down_plots (显示 15 个term)
DMG低甲基化基因KEGG富集分析完成,结果已保存到 ./enrichment_results/DMG/KEGG_down_enrichment.csv
图注:本图展示高甲基化基因(fc > 1,padj < 0.05)的 KEGG 通路富集结果;图元含义见上文「KEGG 功能富集分析可视化图」图注。
# 展示DMG低甲基化基因KEGG富集结果
if os.path.exists(DMG_down_KEGG_file_path):
display_go_plots_html_table(DMG_down_KEGG_dir)