甲基化 + RNA 双组学: 基于 WNN 的单细胞甲基化与转录组双组学整合分析教程
模块简介
本教程介绍如何基于 Scanpy 与 Muon 框架,使用 WNN(Weighted Nearest Neighbors)算法整合单细胞甲基化与转录组(RNA)数据,并完成联合降维、聚类与可视化。
WNN 分析原理
WNN 的核心思想是:不同细胞类型或状态下,不同模态对“区分细胞差异”的贡献可能不同。因此,WNN 会为每个细胞学习一个模态权重,并用该权重融合两个模态的邻居信息。
- 建立单模态邻居图:分别在 RNA 的 PCA 空间与甲基化的 LSI 空间中,为每个细胞寻找 k 近邻(kNN),得到两个模态各自的邻居图。
- 估计细胞特异的模态权重:比较同一细胞在两种模态下的局部邻域一致性与可预测性,为 RNA 与甲基化分别计算权重。直观地说,哪一种模态在该细胞附近提供的结构更稳定、更能解释邻域关系,就会获得更高权重。
- 构建加权融合图并下游分析:将两个单模态邻居图按细胞特异权重融合为 WNN 图。后续可基于该图进行 UMAP 等降维与 Leiden 等聚类。
分析目标
- 联合聚类:融合两类组学的互补信息,更稳定地区分细胞群体。
- 模态贡献评估:在细胞层面评估 RNA 与甲基化对相似性计算的相对贡献。
- 统一可视化:在同一个 UMAP 空间中展示整合后的细胞图谱。
输入文件准备
开始分析前,请准备同一批细胞对应的甲基化与转录组数据:
- 甲基化数据:MCDS 格式数据(通常由 ALLCools 流程生成)。
- RNA 数据:
.rds或.qs格式对象文件(用于导出表达矩阵并在 Scanpy 中读取)。 - Barcode 映射表:DD-MET3 技术需使用
DD-M_bUCB3_whitelist.csv将两个模态的 Barcode 对齐。
import os
import re
import glob
import warnings
import numpy as np
import pandas as pd
import xarray as xr
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
from matplotlib.lines import Line2D
from scipy import sparse
# 生信分析核心库
import scanpy as sc
import scanpy.external as sce
import muon as mu
from muon import atac as ac
from ALLCools.mcds import MCDS
from ALLCools.clustering import (
tsne, significant_pc_test, log_scale, lsi, binarize_matrix,
filter_regions, cluster_enriched_features, ConsensusClustering,
Dendrogram, get_pc_centers, one_vs_rest_dmg
)
from ALLCools.clustering.doublets import MethylScrublet
from ALLCools.plot import *
from harmonypy import run_harmony
from skmisc.loess import loess
# 配置 R 环境(用于 rpy2 调用 R 包)
r_base_path = '/PROJ/home/weiqiuxia/micromamba/envs/muon_env'
os.environ['R_HOME'] = os.path.join(r_base_path, 'lib', 'R')
os.environ['LD_LIBRARY_PATH'] = os.path.join(r_base_path, 'lib', 'R', 'lib') + ':' + os.environ.get('LD_LIBRARY_PATH', '')
r_lib_path = os.path.join(r_base_path, 'lib', 'R', 'library')
os.environ['R_LIBS'] = r_lib_path
os.environ['R_LIBS_USER'] = r_lib_path
os.environ['R_LIBS_SITE'] = r_lib_path
# 全局设置
sc.settings.verbosity = 0
warnings.filterwarnings('ignore')
sc.set_figure_params(dpi=80, figsize=(5, 5))
plt.rcParams['figure.dpi'] = 80
plt.rcParams['savefig.dpi'] = 300
plt.rcParams['figure.figsize'] = (5, 5)
try:
from matplotlib_venn import venn2
HAS_VENN = True
except Exception:
HAS_VENN = False
def mcds_to_adata(mcds_path, var_dim='chrom20k', mc_type='CGN', quant_type='hypo-score'):
"""
将 xarray 格式的 hypo-score 矩阵转换为 AnnData 对象。
参数:
mcds_path: MCDS 文件路径
var_dim: 变量维度,如 'chrom20k'
mc_type: 甲基化类型,如 'CGN'
quant_type: 定量类型,如 'hypo-score'
"""
mcds = MCDS.open(mcds_path, obs_dim='cell', var_dim=var_dim)
adata = mcds.get_score_adata(mc_type=mc_type, quant_type=quant_type, sparse=True)
return adata
def compute_cg_ch_level(df):
"""
计算 CpG 和 CH 的甲基化水平。
参数:
df: 包含 mc 和 cov 列的 DataFrame
"""
cg_cov_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_cov')]
cg_mc_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_mc')]
ch_cov_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_cov')]
ch_mc_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_mc')]
df['CpG_cov'] = df[cg_cov_col].sum(axis=1)
df['CpG_mc'] = df[cg_mc_col].sum(axis=1)
df['CpG%'] = round(df['CpG_mc'] * 100 / df['CpG_cov'], 2)
df['CH_cov'] = df[ch_cov_col].sum(axis=1)
df['CH_mc'] = df[ch_mc_col].sum(axis=1)
df['CH%'] = round(df['CH_mc'] * 100 / df['CH_cov'], 2)
return df
def check_barcode_overlap(rna_adata, met_adata, barcode_map=None):
"""
检查转录组和甲基化数据的细胞 Barcode 重叠情况,并根据共同 Barcode 进行子集提取。
针对 DD-MET3 和 DD-MET5 两种不同技术进行处理。
"""
# 提取原始 Barcode
rna_adata.obs['original_barcode'] = [re.sub('_.*', '', i) for i in rna_adata.obs.index]
met_adata.obs['original_barcode'] = [re.sub('_.*', '', i) for i in met_adata.obs.index]
# 计算初始重叠率
overlap_rate = len(set(rna_adata.obs['original_barcode']) & set(met_adata.obs['original_barcode'])) / met_adata.shape[0]
if overlap_rate < 0.1:
# 处理 DD-MET3 情况:需要通过映射表转换 Barcode
met2rna_barcode = barcode_map[barcode_map['m_cb'].isin(met_adata.obs['original_barcode'])]
rna_adata = rna_adata[rna_adata.obs['original_barcode'].obs.index.isin(met2rna_barcode['gex_cb'])]
met2rna_barcode = met2rna_barcode.set_index('gex_cb')
rna_adata.obs['gex_cb'] = rna_adata.obs.index
rna_adata.obs.index = met2rna_barcode.loc[rna_adata.obs['original_barcode'], 'm_cb']
overlap_rate_test = len(set(rna_adata.obs.index) & set(met_adata.obs.index)) / met_adata.shape[0]
chemistry = 'DD-MET3'
if overlap_rate_test < 0.1:
raise Exception('转录组与甲基化数据的 Barcode 重叠率低于 10%,请检查输入数据。')
else:
# 处理 DD-MET5 情况:Barcode 直接对应
chemistry = 'DD-MET5'
rna_adata.obs.index = rna_adata.obs['original_barcode']
met_adata.obs.index = met_adata.obs['original_barcode']
# 提取共同细胞并同步两个对象
common_index = list(set(rna_adata.obs.index) & set(met_adata.obs.index))
rna_adata = rna_adata[rna_adata.obs.index.isin(common_index)]
rna_adata.obs['chemistry'] = chemistry
met_adata = met_adata[met_adata.obs.index.isin(common_index)]
met_adata.obs['chemistry'] = chemistry
return rna_adata, met_adata, chemistry
def parse_gtf(gtf_file, output_file):
"""
解析 GTF 文件,提取基因类型 (gene_type) 信息。
"""
with open(gtf_file, 'r') as f_in, open(output_file, 'w') as f_out:
for line in f_in:
if line.startswith('#'):
continue
parts = line.strip().split('\t')
if len(parts) < 9:
continue
feature_type = parts[2]
if feature_type != 'gene':
continue
attributes = parts[8]
attr_dict = {}
for attr in attributes.split(';'):
attr = attr.strip()
if not attr:
continue
if ' ' in attr:
key, value = attr.split(' ', 1)
value = value.strip('"')
attr_dict[key] = value
gene_id = attr_dict.get('gene_id', '')
gene_name = attr_dict.get('gene_name', '')
gene_type = attr_dict.get('gene_type', '')
col1 = f"{gene_id}_{gene_name}" if gene_id and gene_name else (gene_id or gene_name)
f_out.write(f"{col1}\t{gene_type}\n")
def rds_to_matrix(filepath, outdir):
"""
调用 R 脚本将 Seurat 对象 (.rds 或 .qs) 转换为 10X 标准矩阵格式。
"""
import rpy2.robjects as robjects
robjects.globalenv['rdsfile'] = filepath
robjects.globalenv['outdir'] = outdir
# 执行 R 代码
robjects.r('''
library(Seurat)
library(qs)
if(endsWith(rdsfile, ".rds")){
obj <- readRDS(rdsfile)
} else {
obj <- qread(rdsfile)
}
dir.create(outdir, recursive = TRUE, showWarnings = FALSE)
# 导出 Counts 矩阵
Matrix::writeMM(GetAssayData(obj, assay = "RNA", layer = "counts"), file = file.path(outdir, "matrix.mtx"))
system(paste0("gzip -f ", file.path(outdir, "matrix.mtx")))
# 导出 Features 信息
features <- data.frame(Feature_id = rownames(obj), Feature_name = rownames(obj), Feature_type = "Gene Expression")
write.table(features, file.path(outdir, "features.tsv"), row.names = F, col.names = F, sep = "\t", quote = F)
system(paste0("gzip -f ", file.path(outdir, "features.tsv")))
# 导出 Barcodes 信息
barcodes <- data.frame(Barcode = colnames(obj))
write.table(barcodes, file.path(outdir, "barcodes.tsv"), row.names = F, col.names = F, sep = "\t", quote = F)
system(paste0("gzip -f ", file.path(outdir, "barcodes.tsv")))
# 导出元数据
meta <- obj@meta.data
write.table(tibble::rownames_to_column(meta, "barcode"), file.path(outdir, "meta.csv"), row.names = FALSE, col.names = TRUE, sep = ",", quote = F)
''')参数设置
输入目录结构说明
重要提示:本流程对输入数据的目录结构有特定要求。程序将根据 indir 参数自动检索 RNA 与甲基化相关的输入文件。建议按照以下结构组织数据:
data/AY1768874914782/ (indir)
├── input.rds # 转录组数据(Seurat 对象或 qs 格式)
└── methylation # 甲基化数据根目录
└── demoWTJW969-task-1 # 任务/批次名称
└── WTJW969 # 样本名称
├── allcools_generate_datasets
│ └── WTJW969.mcds # MCDS 数据集文件夹
│ ├── chrom20k # 20 kb 窗口的甲基化得分矩阵
│ └── ...
└── split_bams
├── filtered_barcode_reads_counts.csv # 过滤后的 Barcode mapped reads 计数
└── WTJW969_cells.csv # 过滤后的细胞质控信息(CpG 数量、覆盖度等)- RNA 数据检索:在
indir路径下匹配.rds或.qs文件。 - 甲基化数据检索:在
indir/methylation路径下检索 MCDS 文件及关联的质控 CSV 文件。
# --------------------------
# 1. 路径与样本设置
# --------------------------
# indir:输入数据目录
# 注意:该目录下应包含转录组文件 (.rds/.qs) 和 methylation 文件夹
# 如果有多个批次的数据位于不同目录,可以用逗号分隔多个路径,例如:'/path/to/data1,/path/to/data2'
indir = '/home/weiqiuxia/workspace/data/AY1768874914782/'
# outdir:分析结果输出目录
# 所有生成的中间文件和最终结果将保存在此目录下
outdir = '/home/weiqiuxia/workspace/project/weiqiuxia/WNN_results'
# sample_col:样本列名
# 用于在 metadata 中区分不同样本,默认 'Sample'。需与 `indir/methylation` 路径下的样本文件夹名称一致
sample_col = 'Sample'
# --------------------------
# 2. 甲基化分析参数
# --------------------------
# var_dim:甲基化特征维度
# 'chrom20k' 表示将基因组划分为 20kb 的窗口进行分析,这是单细胞甲基化常用的分析粒度
var_dim = 'chrom20k'
# total_cpg_number_cutoff:CpG 覆盖度阈值
# 过滤掉检测到的 CpG 位点总数少于此值的低质量细胞
total_cpg_number_cutoff = 500
# dd_met3_barcode_map:DD-MET3 技术专用的 Barcode 映射表
# 如果使用的是 DD-MET3 建库技术,转录组和甲基化的 Barcode 序列不完全一致,需要此表进行转换
dd_met3_barcode_map = '/home/weiqiuxia/workspace/project/weiqiuxia/DD-M_bUCB3_whitelist.csv'
# resolution:聚类分辨率
# 控制聚类的精细程度,数值越大,得到的细胞亚群越多
resolution = 1
# --------------------------
# 3. RNA 分析参数 (可选)
# --------------------------
# rna_meta:外部 RNA 元数据文件路径 (可选)
# 如果有额外的细胞注释信息需要导入,可在此指定 CSV 文件路径
rna_meta = None
# anno_col:细胞注释列名
# 指定 RNA meta数据中用于标记细胞类型的列名
anno_col = 'CellAnnotation'
# reduction_col_prefix:降维坐标列前缀 (可选)
# 若 RNA meta数据中已包含 UMAP 或 t-SNE 坐标 (如 'UMAP_1', 'UMAP_2'),可在此指定前缀
reduction_col_prefix = ''
# 自动创建输出目录
os.makedirs(outdir, exist_ok=True)数据读取
本步骤将分别读取 RNA 与甲基化数据,并根据 Barcode 进行匹配与过滤。
主要流程:
- 加载 Barcode 映射表:用于 DD-MET3 数据的 Barcode 转换。
- RNA 数据转换与加载:自动检测并转换
.qs或.rds文件为 Matrix Market 格式,然后使用 Scanpy 读取。 - 甲基化数据加载与合并:遍历目录读取 MCDS 文件与相关质控信息,并与 RNA 数据按 Barcode 取交集。
# 读取 DD-MET3 Barcode 映射表
# 该表用于解决 RNA 和 Methylation 数据中 Barcode 不一致的问题
if os.path.exists(dd_met3_barcode_map):
dd_met3_barcode_map_df = pd.read_csv(dd_met3_barcode_map, header=0)
else:
print(f"Warning: Barcode map file not found at {dd_met3_barcode_map}. This is only required for DD-MET3 data.")
dd_met3_barcode_map_df = None# 首先获取qs或者rds结尾的文件,并转成矩阵文件,然后使用scanpy读取
for i in os.listdir(indir):
if i.endswith('.qs'):
rds_to_matrix(os.path.join(indir, i), os.path.join(outdir, 'input_matrix'))
breakR callback write-console: Loading required package: sp
R callback write-console:
Attaching package: ‘SeuratObject’
R callback write-console: The following objects are masked from ‘package:base’:
intersect, t
R callback write-console: qs 0.27.3. Announcement: https://github.com/qsbase/qs/issues/103
# scanpy 读取 RNA 数据
adata_rna = sc.read_10x_mtx(f'{outdir}/input_matrix/')
meta = pd.read_csv(f'{outdir}/input_matrix/meta.csv', index_col = 0, header = 0)
adata_rna.obs = meta
adata_rna.obs['CellAnnotation'] = adata_rna.obs['CellAnnotation'].astype('category')#读取甲基化数据,然后与转录组数据取交集,确保甲基化和转录组的数据的细胞一致,barcode一致
keep_col = ['barcode', 'total_cpg_number', 'genome_cov', 'reads_counts', 'CpG%', 'CH%']
obj_met_list = {}
obj_rna_list = {}
indirs = indir.split(',')
samples = []
mcds_paths = {}
sample_path = {}
analysis_batch_dir = os.listdir(os.path.join(indir, 'methylation'))
for i in analysis_batch_dir:
if os.path.isdir(os.path.join(indir, 'methylation', i)):
a_batch_dir = os.path.join(indir, 'methylation', i)
# 获取分析批次下所有样本
samples.extend([ i for i in os.listdir(a_batch_dir)])
for s in samples:
mcds_path = os.path.join(a_batch_dir, f'{s}','allcools_generate_datasets', f'{s}.mcds')
mcds_paths[s] = mcds_path
sample_path[s] = a_batch_dir
obj = mcds_to_adata(mcds_path, var_dim = var_dim)
obj_met_list[s] = obj
# 读取甲基化相关的质控结果
s_cells_meta = pd.read_csv(os.path.join(a_batch_dir, s, 'split_bams', f'{s}_cells.csv'), header = 0)
s_cells_meta['barcode'] = [b.replace('_allc.gz','') for b in s_cells_meta['cell_barcode']]
# 读取每个细胞的mapped reads
s_reads_counts = pd.read_csv(os.path.join(a_batch_dir, s, 'split_bams', 'filtered_barcode_reads_counts.csv'), header = 0)
s_merged_df = s_cells_meta.merge(s_reads_counts, how = 'inner', left_on = 'barcode', right_on = 'barcode')
# 合并以上两个dataframe
s_merged_df = compute_cg_ch_level(s_merged_df)
s_merged_df = s_merged_df[keep_col]
s_merged_df = s_merged_df.set_index('barcode')
# 将甲基化相关的meta添加到甲基化数据中
obj_met_list[s].obs = obj_met_list[s].obs.merge(s_merged_df, left_index = True, right_index = True)
obj_met_list[s] = obj_met_list[s][obj_met_list[s].obs['total_cpg_number'] >= total_cpg_number_cutoff ]
obj_met_list[s].obs['Sample'] = s
# 检查同一个样本的转录组数据和甲基化数据的细胞barcode是否一致,细胞数量是否一致;取交集
adata_rna_s = adata_rna[adata_rna.obs['orig.ident'] == s]
obj_rna_list[s], obj_met_list[s], chemistry = check_barcode_overlap(adata_rna_s, obj_met_list[s], dd_met3_barcode_map_df)
obj_rna_list[s].obs['Sample'] = s
else:
raise Exception('file not found!')# 合并数据
if len(obj_rna_list) > 1:
adata_rna = obj_rna_list[samples[0]].concatenate([obj_rna_list[i] for i in samples[1:] ])
else:
adata_rna = obj_rna_list[samples[0]]
del obj_rna_list
if len(obj_met_list) > 1:
adata_met = obj_met_list[samples[0]].concatenate([obj_met_list[i] for i in samples[1:] ])
else:
adata_met = obj_met_list[samples[0]].copy()
del obj_met_list甲基化数据预处理
单细胞甲基化数据通常具有高维与高稀疏的特点。为更稳定地提取生物学信号,我们采用类似 scATAC-seq 的处理策略:TF-IDF 归一化结合 LSI(Latent Semantic Indexing)降维。
核心步骤说明
本流程使用 ALLCools 计算的 hypo-methylation score(hypo-score)矩阵。该分值范围为 0 到 1,数值越大表示该区域在该细胞中越低甲基化(hypo-methylated)。
- 二值化(Binarization):将 hypo-score 转换为二值信号(0 或 1)。通常将高于阈值且显著的低甲基化区域记为 1,其余记为 0。这样可以降低极端值与技术波动的影响,使模型更关注“是否存在显著的低甲基化信号”。
- 特征过滤(Feature Filtering):去除在极少数细胞中检测到、或在几乎所有细胞中都检测到的基因组区域,保留更有区分度的特征。
- TF-IDF 归一化:
- TF(Term Frequency):校正细胞层面的测序深度差异。
- IDF(Inverse Document Frequency):降低普遍存在特征的权重,提升细胞特异性特征的贡献度。
- LSI 降维:通过奇异值分解(SVD)将高维稀疏矩阵投影到低维潜空间。
- 批次校正(Harmony):若包含多个样本,使用 Harmony 算法在 LSI 空间中校正批次效应。
# 备份原始矩阵
adata_met.layers['raw'] = adata_met.X.copy()
# 1. 数据二值化:hypo-score 越大表示越低甲基化;将高于 cutoff 的值设为 1,其余为 0
binarize_matrix(adata_met, cutoff=0.95)
# 2. 特征过滤:去除覆盖度过低或过高的区域
filter_regions(adata_met)
# 3. TF-IDF 归一化
ac.pp.tfidf(adata_met, scale_factor=1e4)
# 4. LSI 降维 (SVD)
ac.tl.lsi(adata_met)
# 5. 批次校正(多样本时执行)
if len(samples) > 1:
ho = run_harmony(
adata_met.obsm['X_lsi'],
meta_data=adata_met.obs,
vars_use=sample_col,
random_state=0,
max_iter_harmony=20)
adata_met.obsm['X_lsi_harmony'] = ho.Z_corr
met_reduc_use = 'X_lsi_harmony'
else:
met_reduc_use = 'X_lsi'基于甲基化数据的聚类结果
图注:本图展示了基于甲基化模态构建的 UMAP 嵌入。
- 左图:按细胞簇(
leiden)着色。- 右图:按样本(
Sample)着色,用于观察潜在批次效应。注意:甲基化模态的
n_pcs不宜过大;过多成分可能引入噪声并导致过度分群。
met_sig_pcs = significant_pc_test(adata_met, obsm = met_reduc_use, update = False, p_cutoff=0.1)
sc.pp.neighbors(adata_met,use_rep = met_reduc_use, n_neighbors=30, n_pcs = met_sig_pcs,random_state=20)
sc.tl.umap(adata_met, min_dist=.5, random_state=20)
sc.tl.tsne(adata_met,use_rep=met_reduc_use, random_state=20)
sc.tl.leiden(adata_met, resolution=resolution,random_state=20)
warnings.filterwarnings('ignore')
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi=80)
sc.pl.umap(adata_met, color = ['leiden','Sample'], wspace=0.3)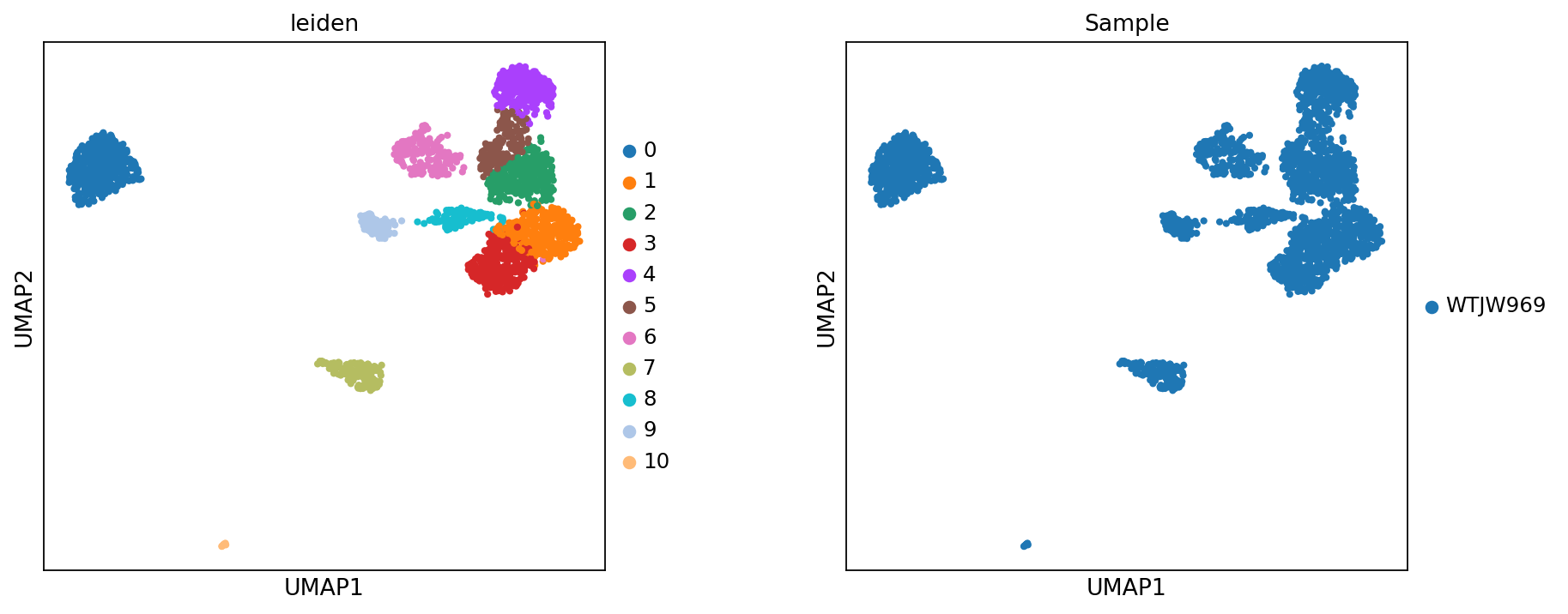
每个细胞的测序深度与甲基化水平展示
图注:本图展示了质控指标在甲基化 UMAP 空间中的分布。
reads_counts:每个细胞的测序深度(mapped reads 数)。CpG%:每个细胞的 CpG 甲基化比例。CH%:每个细胞的 CH 甲基化比例。提示:颜色与数值大小的对应关系以图中颜色条(colorbar)为准。
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi=80)
sc.pl.umap(adata_met, color = ['reads_counts','CpG%','CH%'])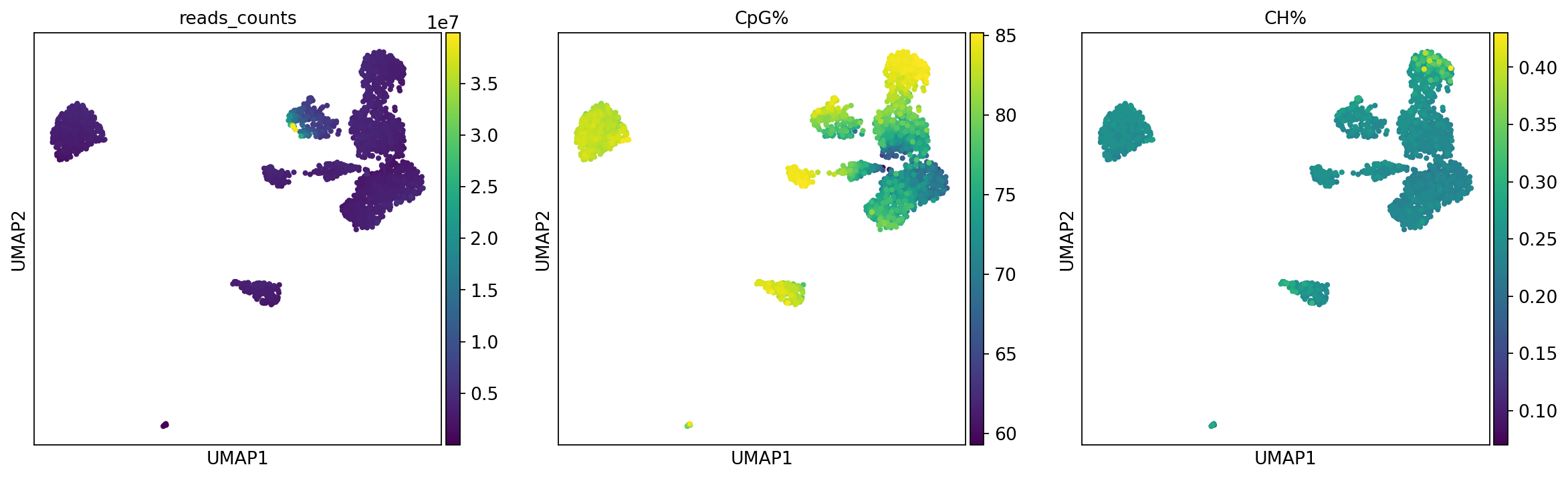
LSI 成分与 QC 指标的相关性分析
在单细胞甲基化数据中,LSI 的第 1 个成分常与测序深度等技术因素相关。为减少技术因素对聚类与降维的影响,建议先检查各 LSI 成分与 reads_counts、CpG% 等指标的相关性,并在需要时在下游建图时跳过相关性过强的成分(常见做法是去掉第 1 个 LSI 成分)。
# 计算 LSI 组件与测序深度 (reads_counts) 和 甲基化水平 (CpG%) 的相关性
lsi_mat = adata_met.obsm[met_reduc_use]
n_components = lsi_mat.shape[1]
components = [f"{met_reduc_use}_{i}" for i in range(n_components)]
# 准备数据
metrics = ['reads_counts', 'CpG%']
cor_data = []
for metric in metrics:
if metric in adata_met.obs.columns:
# 计算每一列 LSI 与 metric 的相关系数
corrs = [np.corrcoef(lsi_mat[:, i], adata_met.obs[metric])[0, 1] for i in range(n_components)]
cor_data.append(corrs)
cor_df = pd.DataFrame(cor_data, index=[m for m in metrics if m in adata_met.obs.columns], columns=components)
# 绘图
fig, ax = plt.subplots(figsize=(11, 3), dpi = 80)
im = ax.imshow(cor_df, cmap='coolwarm', vmin=-1, vmax=1)
ax.set_yticks(np.arange(len(cor_df.index)))
ax.set_yticklabels(cor_df.index)
ax.set_xlabel("LSI Components")
ax.set_title("Correlation between LSI Components and QC Metrics")
plt.colorbar(im, label="Pearson Correlation")
plt.grid(False)
plt.tight_layout()
plt.show()
# 打印与 reads_counts 相关性最高的组件
if 'reads_counts' in cor_df.index:
print("Top LSI components correlated with reads_counts:")
print(cor_df.loc['reads_counts'].abs().sort_values(ascending=False).head())
# 打印与 CpG甲基化 相关性最高的组件
if 'CpG%' in cor_df.index:
print("Top LSI components correlated with CpG%:")
print(cor_df.loc['CpG%'].abs().sort_values(ascending=False).head())
Top LSI components correlated with reads_counts:
X_lsi_0 0.559620
X_lsi_4 0.423008
X_lsi_5 0.397521
X_lsi_2 0.377632
X_lsi_6 0.137376
Name: reads_counts, dtype: float64
Top LSI components correlated with CpG%:
X_lsi_1 0.967854
X_lsi_0 0.284859
X_lsi_2 0.085100
X_lsi_3 0.049610
X_lsi_12 0.041342
Name: CpG%, dtype: float64
RNA 数据预处理
转录组预处理流程
RNA 数据的预处理遵循常用的 Scanpy 流程,目标是去除低质量细胞与低信息基因、减少技术噪声,并保留主要的生物学变异:
- 质量控制(QC):
sc.pp.filter_cells:过滤检测到的基因数过少的细胞(可能为空液滴或低质量细胞)。sc.pp.filter_genes:过滤仅在极少数细胞中表达的基因(信息量较低,可能更多反映噪声)。- 线粒体/核糖体/血红蛋白基因比例:计算这些基因集合的表达占比作为辅助指标;线粒体比例偏高时,常提示细胞受损或应激。
- 归一化(Normalization):
sc.pp.normalize_total:将每个细胞的总计数归一到相同规模(常用 10,000),减小测序深度差异带来的影响。
- 对数变换(Log Transformation):
sc.pp.log1p:进行变换,压缩动态范围并稳定方差,利于后续降维与建图。
- 高变基因选择(Feature Selection):
sc.pp.highly_variable_genes:筛选细胞间变异较大的基因(HVGs),用于后续降维与建图,通常能减少噪声并提升计算效率。
- 数据缩放(Scaling):
sc.pp.scale:将每个基因缩放到均值为 0、方差为 1,避免高表达基因在 PCA 中占据主导。
- PCA 降维(
sc.tl.pca):在 HVG 空间进行主成分分析,提取主要变异方向。 - 批次校正(可选):若包含多个样本,可在 PCA 空间使用 Harmony(
run_harmony)校正批次效应,使同类细胞在不同样本间更好对齐。
adata_rnaobs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'mito', 'Sample', 'raw_Sample', 'resolution.0.5_d30', 'CellAnnotation', 'original_barcode', 'chemistry'
var: 'gene_ids', 'feature_types'
# 1. 过滤低质量细胞和基因
# min_genes=200: 过滤掉检测到的基因数少于 200 的细胞 (可能是死细胞或空液滴)
sc.pp.filter_cells(adata_rna, min_genes=200)
# min_cells=3: 过滤掉在少于 3 个细胞中表达的基因 (可能是测序噪音)
sc.pp.filter_genes(adata_rna, min_cells=3)
# 2. 计算质控指标
# 线粒体基因 (Mitochondrial): 通常以 "MT-" 开头 (人类)
adata_rna.var["mt"] = adata_rna.var_names.str.startswith("MT-")
# 核糖体基因 (Ribosomal): 通常以 "RPS" 或 "RPL" 开头
adata_rna.var["ribo"] = adata_rna.var_names.str.startswith(("RPS", "RPL"))
# 血红蛋白基因 (Hemoglobin): 通常以 "HB" 开头
adata_rna.var["hb"] = adata_rna.var_names.str.contains("^HB[^(P)]")
# 计算 QC 指标,结果将存储在 adata_rna.obs 和 adata_rna.var 中
sc.pp.calculate_qc_metrics(adata_rna, qc_vars=["mt", "ribo", "hb"], inplace=True, log1p=True)# 备份原始 counts 数据,用于差异基因分析等
adata_rna.layers["counts"] = adata_rna.X.copy()
# 1. 归一化 (Normalization)
# 将每个细胞的测序深度标准化 (通常缩放到 10,000 reads)
sc.pp.normalize_total(adata_rna, target_sum=1e4)
# 2. 对数变换 (Log Transformation)
# log(x+1) 变换,稳定方差并使数据接近正态分布
sc.pp.log1p(adata_rna)
# 3. 高变基因选择 (Feature Selection)
# 筛选出在细胞间表达差异显著的基因 (HVGs),这里选取前 2000 个
sc.pp.highly_variable_genes(adata_rna, flavor='seurat_v3', n_top_genes=2000, batch_key="Sample")
# 4. 数据缩放 (Scaling)
# 将每个基因的表达量缩放到均值为 0,方差为 1,确保 PCA 不受高表达基因主导
sc.pp.scale(adata_rna, max_value=10)
# 5. PCA 降维
# 在 HVG 空间进行主成分分析,提取主要变异轴
sc.tl.pca(adata_rna, svd_solver='arpack')# 6. 批次校正 (Harmony Integration)
# 仅当存在多个样本时才进行批次校正
if len(samples) > 1:
print("Running Harmony for batch correction...")
ho = run_harmony(
adata_rna.obsm['X_pca'],
meta_data=adata_rna.obs,
vars_use='Sample',
random_state=0,
max_iter_harmony=20
)
# 将校正后的 PCA 坐标存储在 obsm 中
adata_rna.obsm['X_pca_harmony'] = ho.Z_corr
rna_reduc_use = 'X_pca_harmony'
else:
print("Only one sample detected, skipping batch correction.")
rna_reduc_use = 'X_pca'
# 7. 计算邻居图 (Neighbors Graph)
# 基于选择的降维结果 (X_pca 或 X_pca_harmony) 计算细胞间的邻居关系
sc.pp.neighbors(adata_rna, use_rep=rna_reduc_use, n_neighbors=15, n_pcs=30, random_state=20)
# 8. UMAP 降维与聚类
# UMAP 用于可视化,Leiden 用于聚类
sc.tl.umap(adata_rna, random_state=20)
# sc.tl.tsne(adata_rna, use_rep=rna_reduc_use, random_state=20) # 可选 t-SNE
sc.tl.leiden(adata_rna, resolution=resolution, random_state=20)基于转录组数据的聚类结果
- 若不同样本在 UMAP 上明显分离,通常提示存在批次效应;Harmony 往往可以改善。
- 若样本本身已经充分混合,Harmony 的影响可能较小。
注意:Harmony 主要用于校正样本/批次差异;如果上游 QC、过滤或归一化不充分,或某些样本质量显著较差,批次校正效果会受限。
图注:RNA UMAP(细胞分群与样本)。本图展示 RNA 模态在 UMAP 空间中的分布,分别按
leiden(细胞分群)与Sample(样本)着色。
- 坐标轴:UMAP1 与 UMAP2,为 UMAP 降维后的两个坐标。
- 颜色(
leiden):不同颜色代表不同细胞簇,用于观察分群结构。- 颜色(
Sample):不同颜色代表不同样本,用于评估批次效应与样本组成差异。- 点:每个点代表一个细胞。
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi=80)
sc.pl.umap(adata_rna, color = ['leiden','Sample'], wspace=0.3)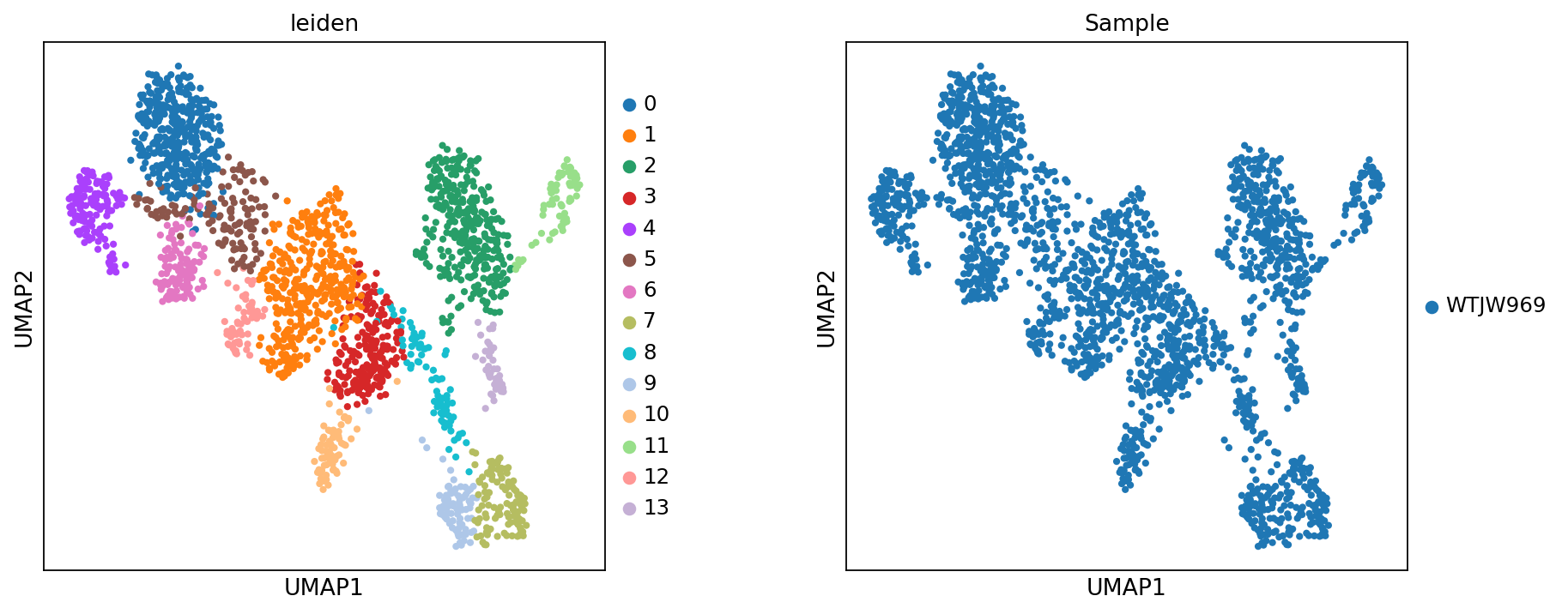
图注:RNA UMAP(标志基因表达)。本图展示 RNA 模态在 UMAP 空间中标志基因的表达分布。
- 坐标轴:UMAP1 与 UMAP2,为 UMAP 降维后的两个坐标。
- 颜色:颜色深浅表示表达量高低,颜色越深表示表达量越高。
- 点:每个点代表一个细胞。
提示:示例标志基因包括 CD3D、THEMIS(T 细胞)、NKG7(NK 细胞)、CD8A(CD8+ T 细胞)、CD4(CD4+ T 细胞)、CD79A 与 MS4A1(B 细胞)、JCHAIN(浆细胞)、LYZ(单核细胞)、LILRA4(pDC 细胞)。
colors = ["#F5E6CC", "#FDBB84", "#E34A33", "#7F0000"]
my_warm_cmap = mcolors.LinearSegmentedColormap.from_list("white_orange_red", colors, N=256)
marker_names = ["CD3D", "THEMIS", "NKG7", "CD8A", "CD4", "IL7R", "S100A4", "CCR7",
"CD79A","MS4A1","JCHAIN",
"LYZ","FCN1","FCGR3A","CST3","MKI67"]
sc.pl.umap(
adata_rna,
color=marker_names,
ncols=4,
color_map=my_warm_cmap,
vmax='p99',
s=10,
frameon=False,
vmin=0,
show=False
)
plt.gcf().set_size_inches(12, 8)
plt.show()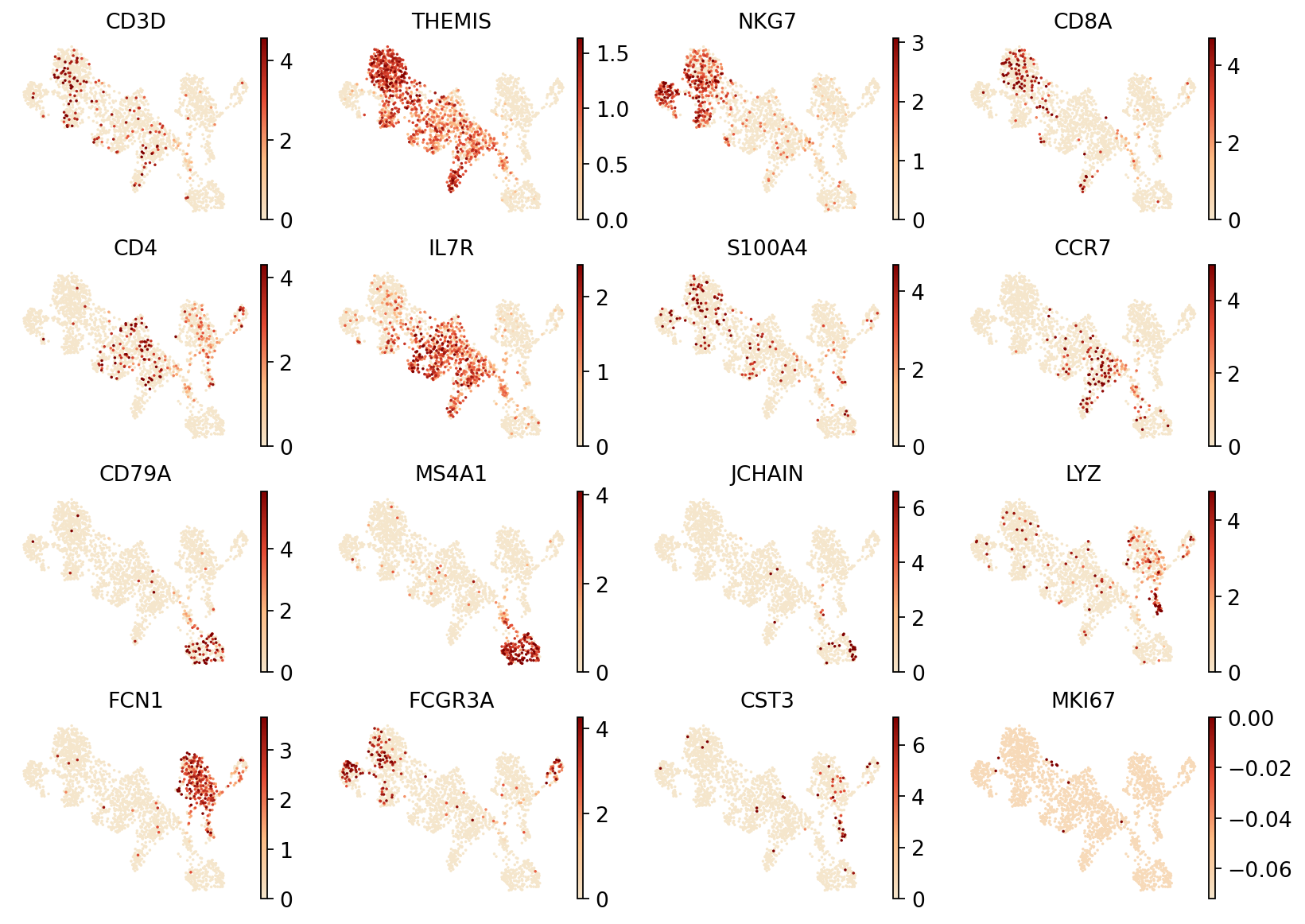
celltype = {
'0': 'CD8+ T',
'1': 'Navie CD4+ T',
'2': 'CD14+ Mono',
'3': 'Memory CD4+ T',
'4': 'NK',
'5': 'T',
'6': 'T',
'7': 'B',
'8': 'Multiplet',
'9': 'B',
'10': 'Memory CD4+ T',
'11': 'FCGR3A+ Mono',
'12': 'Navie CD4+ T',
'13': 'DC'
}
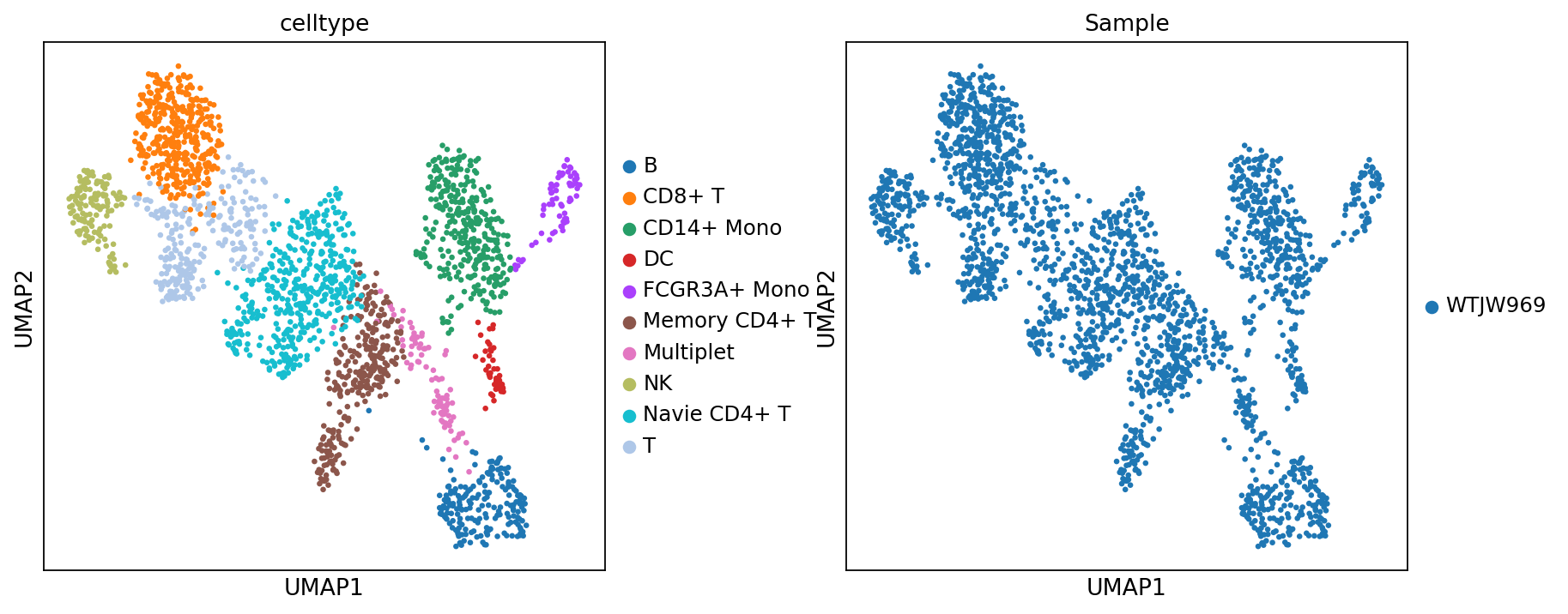
adata_rna.obs['celltype'] = adata_rna.obs['leiden'].map(celltype)图注:RNA UMAP(细胞类型与样本)。本图展示 RNA 模态在 UMAP 空间中的分布,分别按
celltype(细胞类型)与Sample(样本)着色。
- 坐标轴:UMAP1 与 UMAP2,为 UMAP 降维后的两个坐标。
- 颜色(
celltype):不同颜色代表不同细胞类型,用于查看注释结果是否与分群结构一致。- 颜色(
Sample):不同颜色代表不同样本,用于评估批次效应与样本组成差异。- 点:每个点代表一个细胞。
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi = 80)
sc.pl.umap(
adata_rna,
color=['celltype', 'Sample'],
ncols=2,
s=35,
wspace=0.3
)
WNN 多组学整合分析
WNN 原理与步骤
本节将把 RNA 与甲基化两个模态放入同一个 MuData 容器,并用 WNN(Weighted Nearest Neighbors)构建联合邻居图,用于后续的联合降维与聚类。
核心步骤:
- 构建多组学容器:使用
mu.MuData将预处理后的 RNA(mdata['rna'])与甲基化(mdata['methy'])对象封装在一起。 - 计算单模态邻居:
- RNA:使用 PCA 表征(
X_pca或X_pca_harmony)计算邻居。 - 甲基化:使用 LSI 表征(
X_lsi或X_lsi_harmony)计算邻居。
- RNA:使用 PCA 表征(
- WNN 融合(
mu.pp.neighbors):- 对每个细胞,算法会比较两种模态在其局部邻域上的一致性,并据此学习细胞特异的模态权重。
- 然后按权重融合两种模态的邻居信息,生成以
key_added='wnn'命名的一组邻居图结果(通常写入mdata.uns['wnn'],并在mdata.obsp中保存对应的邻接矩阵)。
- 联合降维与聚类:
- UMAP(
mu.tl.umap):基于 WNN 图生成统一的二维嵌入坐标。 - Leiden 聚类(
mu.tl.leiden或sc.tl.leiden):基于 WNN 图识别细胞簇。
- UMAP(
WNN 的优势在于能同时利用两种模态的互补信息:当某些细胞在 RNA 上更容易区分时,RNA 权重会更高;当某些差异更多体现在表观层面时,甲基化权重会更高。
# 构建多组学 MuData 对象
mdata = mu.MuData({
'rna': adata_rna,
'methy': adata_met})确定各模态的有效降维成分数量
在构建 WNN 邻居图之前,需要先确定每个模态使用多少个降维成分:
- RNA:通常对应 PCA 的主成分数量。
- 甲基化:通常对应 LSI 的成分数量。
本教程使用 significant_pc_test 对降维成分进行一个“有效性评估”,帮助我们选择更可能包含生物学信号的维度数量。
p_cutoff=0.1:显著性阈值。
rna_sig_pcs = significant_pc_test(adata_rna, obsm=rna_reduc_use, update=False, p_cutoff=0.1)
met_sig_pcs = significant_pc_test(adata_met, obsm=met_reduc_use, update=False, p_cutoff=0.1)
print(f"Significant PCs for RNA: {rna_sig_pcs}")
print(f"Significant LSI components for Methylation: {met_sig_pcs}")11 components passed P cutoff of 0.1.
Significant PCs for RNA: 12
Significant LSI components for Methylation: 11
运行 WNN 分析
本步骤将分别在两个模态上计算邻居图,再基于 WNN 融合得到联合邻居图。
- 使用上一步得到的有效成分数量(
n_pcs),分别为 RNA(PCA)与甲基化(LSI)计算邻居。 - 再调用
mu.pp.neighbors构建 WNN 图,并在此基础上运行 UMAP 与 Leiden 聚类。
sc.pp.neighbors(mdata['rna'], use_rep=rna_reduc_use, n_pcs=rna_sig_pcs)
sc.pp.neighbors(mdata['methy'], use_rep=met_reduc_use, n_pcs=met_sig_pcs)
# 2. 计算 WNN 联合邻居图并估计模态权重
# 这一步会写入以 key_added='wnn' 命名的一组邻居图结果(常见为 mdata.uns['wnn'],以及 mdata.obsp['wnn_connectivities']/mdata.obsp['wnn_distances'])
# 同时为每个细胞添加模态权重信息(列名通常带有模态前缀,例如 rna:mod_weight、methy:mod_weight)
mu.pp.neighbors(mdata, key_added='wnn', add_weights_to_modalities=True, random_state=20)
# 3. 取两个模态细胞的交集(确保分析对象一致)
# 这一步会过滤掉只在单一模态中存在的细胞
mu.pp.intersect_obs(mdata)
# 4. 基于 WNN 图进行 UMAP 降维与聚类(Leiden)
mu.tl.umap(mdata, neighbors_key='wnn', random_state=20)
sc.tl.leiden(mdata, neighbors_key='wnn', random_state=20)mdata.obsm["X_wnn_umap"] = mdata.obsm["X_umap"]sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi = 80)
mu.pl.embedding(mdata, basis="X_wnn_umap", color=["rna:mod_weight", "methy:mod_weight"], wspace=0.3)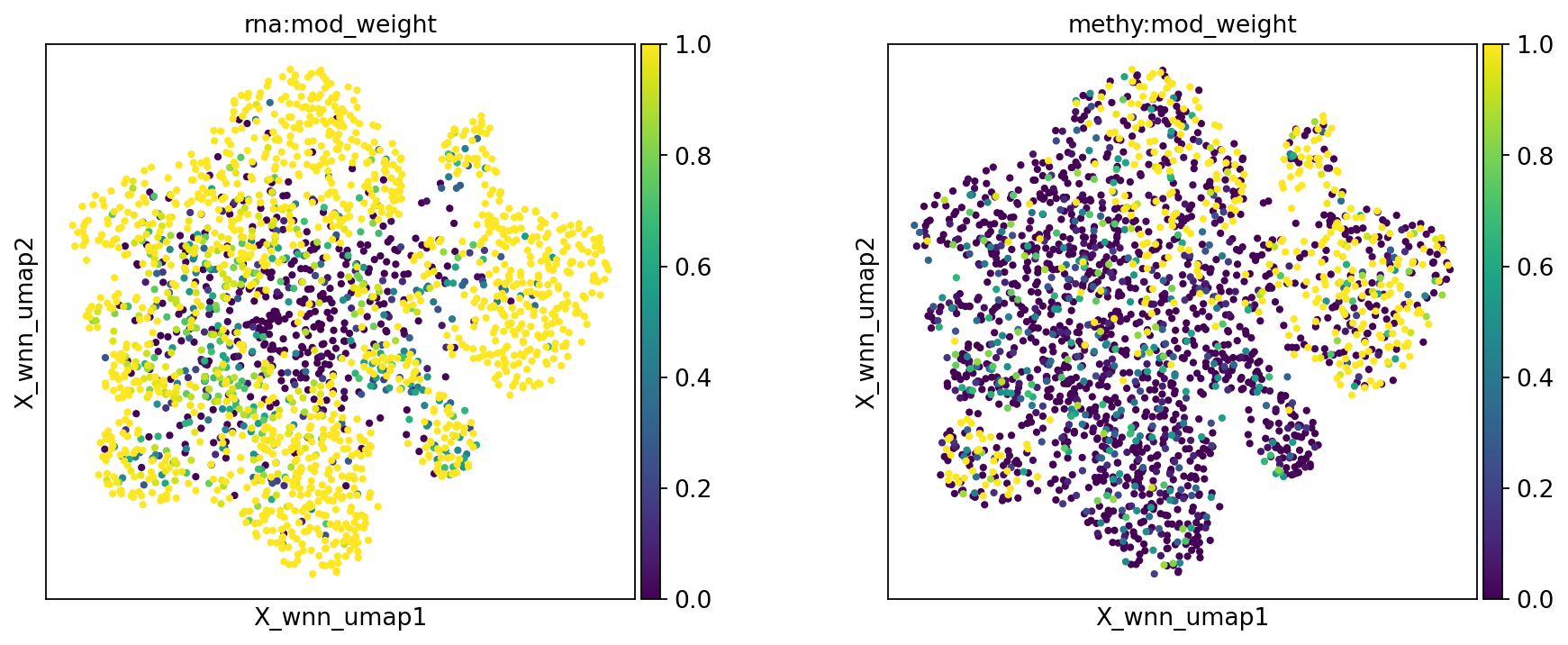
基于 WNN 的降维与聚类结果展示
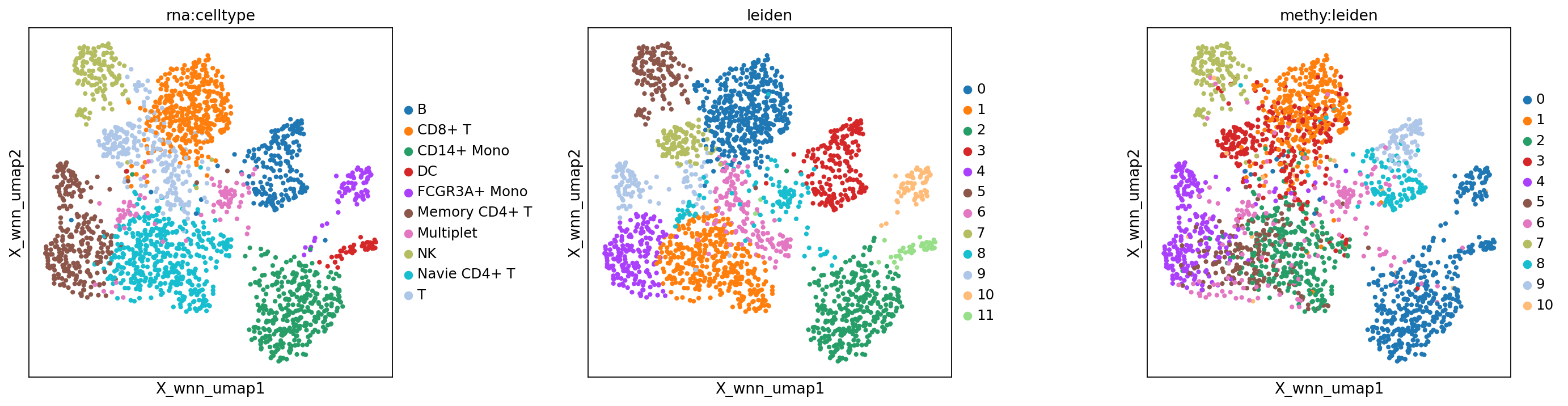
图注:本图对比不同模态下的聚类/注释结果。
- 左图:RNA 单模态细胞类型注释(
rna:celltype)。- 中图:WNN 联合聚类结果(
wnn:leiden)。- 右图:甲基化单模态聚类结果(
methy:leiden)。
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi = 80)
mu.pl.embedding(mdata, basis="X_wnn_umap", color=["rna:celltype", "leiden", "methy:leiden"], wspace=0.4)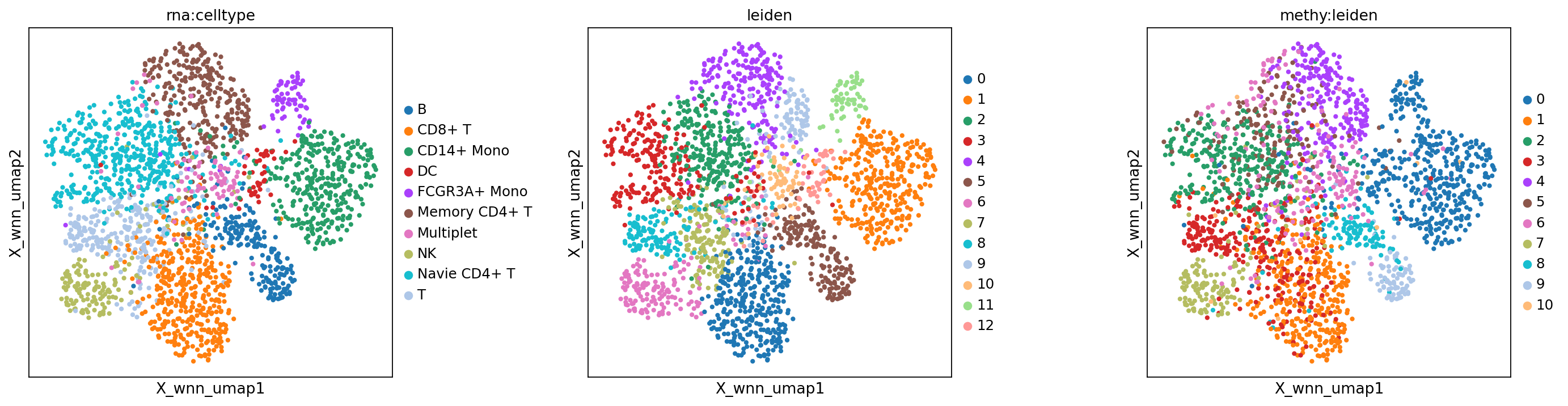
优化参数(可选)
如果初次分析中发现分群效果不理想(例如不同类型细胞混在一起,或已知亚群没有分开),可能是用于建图的成分数量偏少,导致部分生物学差异没有被捕捉到。
此时可以尝试适度增加 n_pcs(RNA 的 PCA 成分数或甲基化的 LSI 成分数),并重新构建 WNN 图,再运行 UMAP 与聚类。
提示:成分数量也不宜过大;过多成分可能引入噪声并导致过度分群。
# 手动指定更多的 PCs/LSI 组件以提高分辨率
# 注意:n_pcs 数量不应超过总的主成分/LSI 组件数量 (通常为 50 或 30)
print("Re-running WNN with manually adjusted n_pcs...")
sc.pp.neighbors(mdata['rna'], use_rep=rna_reduc_use, n_pcs=20) # RNA 增加到 20
sc.pp.neighbors(mdata['methy'], use_rep=met_reduc_use, n_pcs=50) # Methylation 增加到 50 (最大值)
# 重新计算 WNN 图
mu.pp.neighbors(mdata, key_added='wnn', add_weights_to_modalities=True, random_state=20)
mu.pp.intersect_obs(mdata)
# 重新运行 UMAP 和聚类
mu.tl.umap(mdata, neighbors_key='wnn', random_state=20)
sc.tl.leiden(mdata, neighbors_key='wnn', random_state=20)
# 保存新的 UMAP 坐标
mdata.obsm["X_wnn_umap"] = mdata.obsm["X_umap"]
# 可视化新的结果
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi=80)
mu.pl.embedding(mdata, basis="X_wnn_umap", color=["rna:mod_weight", "methy:mod_weight"], wspace=0.3)
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(5, 5), dpi=80)
mu.pl.embedding(mdata, basis="X_wnn_umap", color=["rna:celltype", "leiden", "methy:leiden"], wspace=0.4)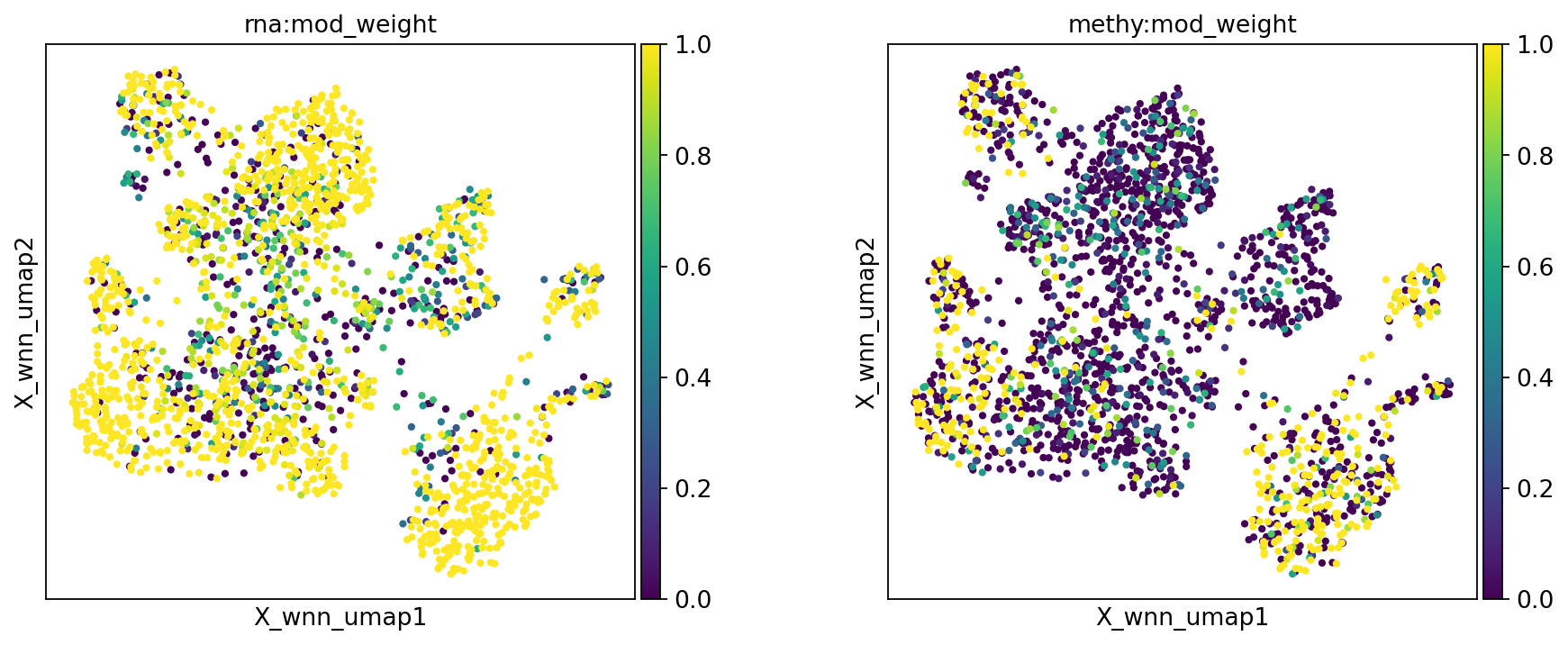
结果保存与输出
分析完成后,将处理好的数据与结果保存为标准格式,便于后续复用或下游分析。
保存的文件包括:
adata_rna.h5ad:RNA 单模态数据对象,包含预处理、降维、聚类及注释信息。adata_met.h5ad:甲基化单模态数据对象,包含预处理、降维及聚类信息。mdata.h5mu:多组学 MuData 对象,包含两个模态及 WNN 整合分析结果。
注意:
.h5mu与.h5ad文件保存了完整的分析状态,载入后可直接进行可视化或下游分析。- 文件可能较大,请确保输出目录有足够的存储空间。
adata_rna.write_h5ad(f'{outdir}/adata_rna.h5ad')
adata_met.write_h5ad(f'{outdir}/adata_met.h5ad')
mdata.write(f'{outdir}/mdata.h5mu')