单细胞甲基化双细胞识别教程 (ALLCools / MethylScrublet)
时长: 4 分钟
字数: 772 字
更新: 2026-02-28
阅读: 0 次
加载 Python 包
python
import os
import re
import glob
from ALLCools.mcds import MCDS
from ALLCools.clustering import tsne, significant_pc_test, log_scale, lsi, binarize_matrix, filter_regions, cluster_enriched_features, ConsensusClustering, Dendrogram, get_pc_centers
from ALLCools.clustering.doublets import MethylScrublet
from ALLCools.plot import *
import scanpy as sc
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
from matplotlib.lines import Line2D
import warnings
import xarray as xr
from ALLCools.clustering import one_vs_rest_dmg
import pybedtools
from scipy import sparsepython
load = True
mc_type = 'CGN'
# Clustering resolution
n_neighbors = 10
expected_doublet_rate=0.06
plot_type = 'static'
mcds_list = []
cell_number = []
samples = ["HC10_12","HC14_21"]单细胞甲基化多样本 MCDS 合并
python
adata_met = sc.read_h5ad("adata_met.h5ad")
for i in samples:
keep_barcodes = [ re.sub('\\-.*','',b) for b in adata_met.obs[adata_met.obs["Sample"] == i].index ]
mcds = MCDS.open(os.path.join(f'{i}', f'{i}_methy','step3','allcools_generate_datasets', f'{i}.mcds'), obs_dim = 'cell', var_dim = 'chrom1M', use_obs = keep_barcodes)
suffix = samples.index(i)
if len(samples) > 1:
mcds = mcds.assign_coords(cell=[ f'{i}-{suffix}' for i in mcds.cell.values ])
mcds_list.append(mcds)
cell_number += [i]*len(mcds.cell.values)
if len(samples) > 1:
combined = xr.concat(mcds_list, dim='cell')
else:
combined = mcds_list[0]
combined = combined.assign_coords(cell = adata_met.obs.index)
mc = combined[f'chrom1M_da'].sel({
'count_type': 'mc'
})
cov = combined[f'chrom1M_da'].sel({
'count_type': 'cov'
})
if load and (combined.get_index('cell').size <= 20000):
mc.load()
cov.load()单细胞甲基化数据的双细胞识别
双细胞是指在单细胞测序过程中,两个或多个细胞意外粘连并被作为一个“细胞”进行测序的技术伪影。双细胞会引入混合的表达/甲基化特征,严重干扰后续的细胞分群和差异分析(例如,可能错误地将双细胞鉴定为新的中间态细胞类型)。
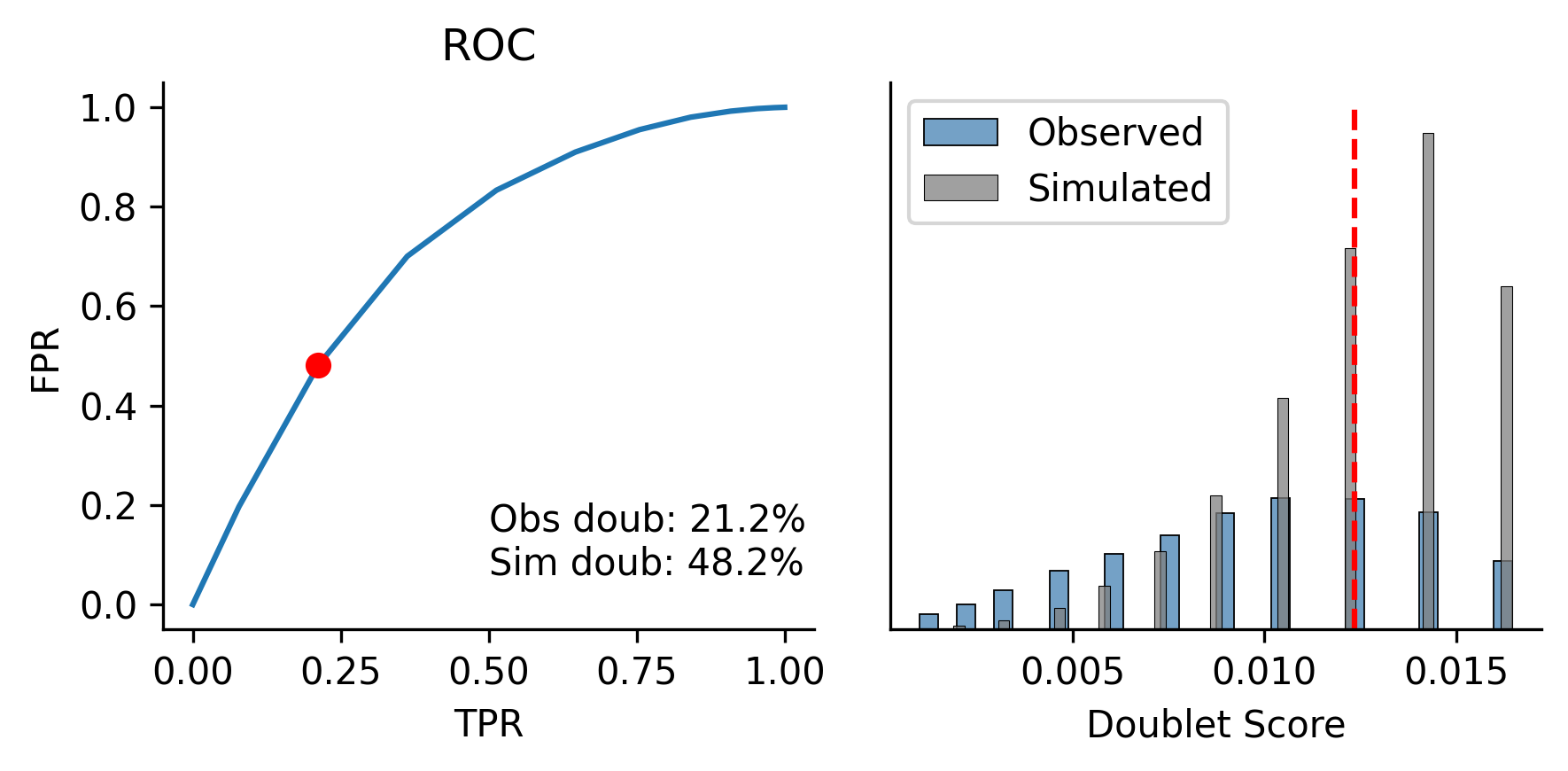
使用 MethylScrublet 算法来识别潜在的双细胞。该算法通过模拟人造双细胞,并将其与观测数据进行比对,从而计算每个细胞的“双细胞评分”。
python
scrublet = MethylScrublet(sim_doublet_ratio=2.0,
n_neighbors=n_neighbors,
expected_doublet_rate=expected_doublet_rate,
stdev_doublet_rate=0.02,
metric='euclidean',
random_state=0,
n_jobs=-1)
score, judge = scrublet.fit(mc, cov, clusters=adata_met.obs["celltype"])
adata_met.obs['met_doublet_score'] = score
adata_met.obs['met_is_doublet'] = judge
scrublet.plot()
adata_met.obs['met_is_doublet'] = adata_met.obs['met_is_doublet'].astype('category')output
Calculating mC frac of observations...n Simulating doublets...n PCA...n Calculating doublet scores...n Automatically set threshold to 0.01
Detected doublet rate = 21.2%
Estimated detectable doublet fraction = 48.2%
Overall doublet rate:
Expected = 6.0%
Estimated = 44.1%
Detected doublet rate = 21.2%
Estimated detectable doublet fraction = 48.2%
Overall doublet rate:
Expected = 6.0%
Estimated = 44.1%
python
plt.rcParams['figure.dpi'] = 150
plt.rcParams['figure.figsize'] = (3,3)
sc.pl.umap(adata_met,
color = ['met_doublet_score', 'met_is_doublet'],
ncols = 2)output
/PROJ2/FLOAT/jinwen/apps/miniconda3/envs/allcools/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:394: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
cax = scatter(
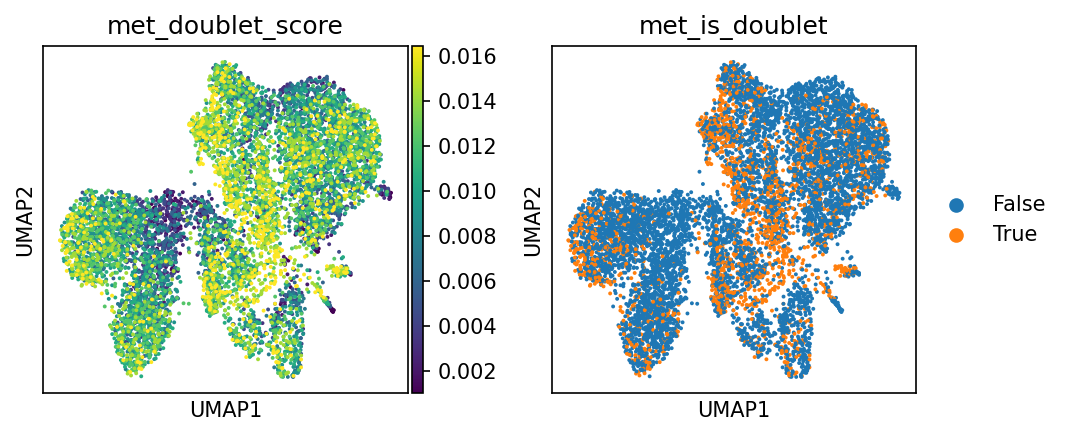
双细胞评分与预测结果
- 左图 (met_doublet_score):双细胞评分。颜色越亮(黄色),表示该细胞与模拟的双细胞特征越相似,是双细胞的可能性越高。
- 右图 (met_is_doublet):双细胞判定结果。
- 橙色点 (True):被判定为双细胞(Doublet)。
- 蓝色点 (False):判定为正常单细胞(Singlet)。