甲基化 + RNA 双组学: 基础分析流程
模块简介
本模块专为单细胞甲基化双组学数据(同时包含 RNA 和甲基化信息)的基础分析而设计。
本模块实现了以下核心分析流程:
- 数据整合 (Data Integration):整合多个样本的单细胞 RNA 测序和单细胞甲基化测序数据。
- 质量控制 (Quality Control):对 RNA 和甲基化数据进行独立的质控过滤,确保数据质量。
- 批次效应校正 (Batch Effect Correction):使用 Harmony 算法校正多样本间的技术变异,保留生物学信号。
- 细胞图谱构建 (Cell Atlas Construction):通过降维、聚类和可视化,构建单细胞图谱,识别细胞类型和亚群。
输入文件准备
本模块需要以下输入文件:
必需文件
- 单细胞 RNA 测序数据:基因表达矩阵(
filtered_feature_bc_matrix目录)。 - 单细胞甲基化数据:MCDS 格式的甲基化数据集(
.mcds文件)。 - Barcode 映射文件(可选):如果使用 DD-MET3 试剂盒,用于关联 RNA 和甲基化数据的细胞 Barcode 对应关系。
文件结构示例
data/
├── AY1768874914782
│ └── methylation
│ └── demoWTJW969-task-1
│ └── WTJW969
│ ├── allcools_generate_datasets
│ │ └── WTJW969.mcds # 甲基化数据
│ ├── filtered_feature_bc_matrix # 转录组表达矩阵
│ └── split_bams
│ ├── WTJW969_cells.csv
│ └── filtered_barcode_reads_counts.csv
└── AY1768876253533
└── methylation
└── demo4WTJW880-task-1
└── WTJW880
├── allcools_generate_datasets
│ └── WTJW880.mcds # 甲基化数据
├── filtered_feature_bc_matrix # 转录组表达矩阵
└── split_bams
├── WTJW880_cells.csv
└── filtered_barcode_reads_counts.csv
DD-M_bUCB3_whitelist.csv # Barcode 映射文件(可选)WARNING
本教程适用于上述目录结构。如果您的文件组织结构不同,请相应调整文件夹路径或修改代码中的文件地址。
# --- 导入必要的 Python 包 ---
# 系统操作和文件处理
import os
import re
import glob
import warnings
# ALLCools 相关包:用于甲基化数据处理
from ALLCools.mcds import MCDS
from ALLCools.clustering import (
tsne, significant_pc_test, log_scale, lsi,
binarize_matrix, filter_regions, cluster_enriched_features,
ConsensusClustering, Dendrogram, get_pc_centers, one_vs_rest_dmg
)
from ALLCools.clustering.doublets import MethylScrublet
from ALLCools.plot import *
# 单细胞分析工具
import scanpy as sc
import scanpy.external as sce
# 批次效应校正
from harmonypy import run_harmony
# 数据处理和可视化
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
from matplotlib.lines import Line2D
# 其他工具
import xarray as xr
import pybedtools
from scipy import sparse# --- 输入参数配置 ---
## samples:待分析的样本列表
samples = ["WTJW969", "WTJW880"]
## 文件路径配置:根据新的文件层级结构配置
## 格式:{样本名: {'top_dir': 顶层目录, 'demo_dir': demo目录}}
## 示例:{"WTJW969": {"top_dir": "AY1768874914782", "demo_dir": "demoWTJW969-task-1"}}
sample_path_config = {
"WTJW969": {"top_dir": "AY1768874914782", "demo_dir": "demoWTJW969-task-1"},
"WTJW880": {"top_dir": "AY1768876253533", "demo_dir": "demo4WTJW880-task-1"}
}
## 数据根目录(默认为 "data")
data_root = "data"
## reagent_type:试剂盒类型('DD-MET3' 或 'DD-MET5'),当所有样本一致时可仅写此处
## reagent_type_map:按样本配置试剂类型(可选)。若存在,则优先于 reagent_type
## 例如 reagent_type_map = {"样本A": "DD-MET3", "样本B": "DD-MET5"}
## DD-MET3:RNA 和甲基化使用不同 Barcode,需通过映射文件关联
## DD-MET5:RNA 和甲基化使用相同 Barcode,不需要映射
reagent_type = "DD-MET3"
reagent_type_map = {"WTJW969": "DD-MET5", "WTJW880": "DD-MET3"}
## var_dim:甲基化数据的基因组窗口大小(分辨率)
var_dim = 'chrom20k'
## obs_dim:观测维度(通常是 'cell')
obs_dim = 'cell'
## mc_type:甲基化位点类型('CGN' 表示 CpG 位点)
mc_type = 'CGN'
## quant_type:甲基化评分的量化方式('hypo-score' 表示低甲基化评分)
quant_type = 'hypo-score'
## 画图色板
my_palette = [
"#66C2A5", "#FC8D62", "#8DA0CB", "#E78AC3",
"#A6D854", "#FFD92F", "#E5C494", "#B3B3B3",
"#8DD3C7", "#BEBADA", "#FB8072", "#80B1D3",
"#FDB462", "#B3DE69", "#FCCDE5", "#BC80BD",
"#CCEBC5", "#FFED6F", "#A6CEE3", "#FB9A99"
]数据质控与整合
本节首先对 RNA 和甲基化数据分别进行质控过滤,然后计算两个数据集的 barcode 交集,确保后续分析的细胞一致性。
RNA 数据整合与质控过滤
本节对多个样本的 RNA 数据进行整合,并进行质控过滤,去除低质量细胞和基因,确保后续分析的可靠性。
质控过滤策略:
- n_genes_by_counts(检测到的基因数):过滤基因数 < 200 或 > 10000 的细胞。基因数过少的细胞可能捕获效率低或质量差;基因数过多的细胞可能是双细胞 (doublet) 或技术异常。
- total_counts(总 UMI 数):过滤总 UMI 数 < 200 或 > 30000 的细胞。UMI 数过少的细胞测序深度不足,数据不可靠;UMI 数过多的细胞可能是双细胞或技术异常。
- 线粒体基因比例:过滤线粒体基因比例 > 20% 的细胞。线粒体基因比例过高的细胞可能已死亡或受损,属于低质量细胞。
- 低表达基因过滤:过滤在少于 3 个细胞中表达的基因。这些基因表达量极低,可能是噪声或测序错误,对分析贡献有限。
质控过滤后,将输出质控前后的细胞数和基因数统计信息,便于评估过滤效果。
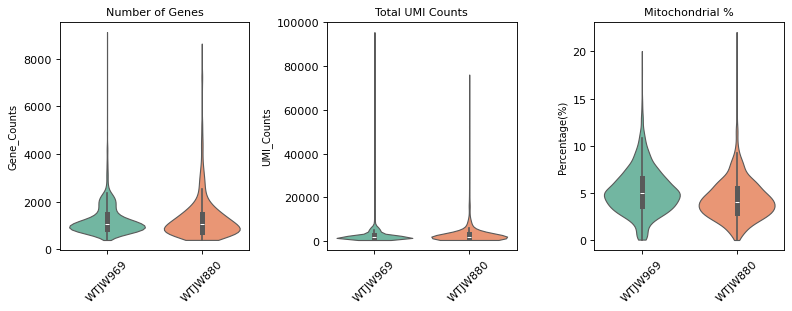
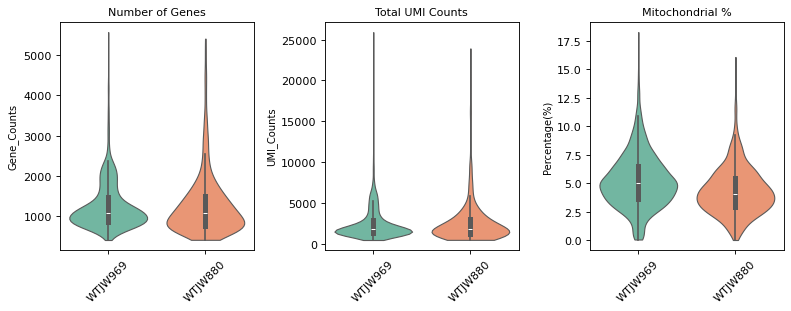
图注(RNA 质控小提琴图):
下图展示了 RNA 数据质控过滤前后质控指标的分布情况,按样本 (Sample) 分组显示。
- 质控前小提琴图:显示质控过滤前的质控指标分布,包括
n_genes_by_counts(检测到的基因数)、total_counts(总 UMI 数)和pct_counts_mt(线粒体基因比例)。可用于评估原始数据质量和识别异常值。- 质控后小提琴图:显示质控过滤后的质控指标分布。过滤后,低质量细胞(基因数过少/过多、UMI 数异常、线粒体比例过高)已被去除,数据质量得到提升。
- 横坐标:不同样本 (Sample),便于比较不同样本间的质控指标差异。
- 纵坐标:各质控指标的数值。小提琴图的宽度表示该数值区间的细胞密度,越宽表示该区间的细胞越多。
- 用途:通过对比过滤前后的分布,可以评估质控过滤的效果,确认过滤阈值是否合理。
warnings.filterwarnings('ignore')
# --- RNA 多样本数据整合与质控过滤 ---
# 读取各样本的 RNA 表达数据
obj_rna_list = {}
for i in samples:
# 根据新的文件层级结构构建路径:../../data/{top_dir}/methylation/{demo_dir}/{sample}/filtered_feature_bc_matrix
top_dir = sample_path_config[i]['top_dir']
demo_dir = sample_path_config[i]['demo_dir']
s_rna_path = os.path.join('../../',data_root, top_dir, 'methylation', demo_dir, i, 'filtered_feature_bc_matrix')
obj_rna_list[i] = sc.read_10x_mtx(s_rna_path)
# 合并所有样本的数据
adata_rna = obj_rna_list[samples[0]].concatenate([obj_rna_list[i] for i in samples[1:]])
# 添加样本信息
adata_rna.obs['Sample'] = adata_rna.obs['batch'].map(
{f'{index}': value for index, value in enumerate(samples)}
)
# --- 质控过滤 ---
# 计算质控指标
adata_rna.var['mt'] = adata_rna.var_names.str.startswith('MT-') # 线粒体基因标记
sc.pp.calculate_qc_metrics(adata_rna, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
# 保存质控前的统计信息
n_cells_before = adata_rna.n_obs
n_genes_before = adata_rna.n_vars
print(f"质控前细胞数: {n_cells_before}")
print(f"质控前基因数: {n_genes_before}")
# --- 质控前小提琴图可视化 ---
print("\n=== 质控前质控指标分布 ===")
# sc.pl.violin(adata_rna, keys=['n_genes_by_counts', 'total_counts', 'pct_counts_mt'],
# groupby='Sample', rotation=45, multi_panel=True)
fig, (ax1, ax2, ax3) = plt.subplots(1, 3, figsize=(10, 4), dpi=80)
sc.pl.violin(adata_rna, keys='n_genes_by_counts', groupby='Sample', rotation=45, ax=ax1, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax1.set_title("Number of Genes", fontsize=10)
ax1.set_ylabel("Gene_Counts", fontsize=9)
ax1.set_xlabel("")
sc.pl.violin(adata_rna, keys='total_counts', groupby='Sample', rotation=45, ax=ax2, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax2.set_title("Total UMI Counts", fontsize=10)
ax2.set_ylabel("UMI_Counts", fontsize=9)
ax2.set_xlabel("")
sc.pl.violin(adata_rna, keys='pct_counts_mt', groupby='Sample', rotation=45, ax=ax3, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax3.set_title("Mitochondrial %", fontsize=10)
ax3.set_ylabel("Percentage(%)", fontsize=9)
ax3.set_xlabel("")
plt.tight_layout()
plt.show()
# 过滤低质量细胞
# nFeature (n_genes_by_counts): 基于每个细胞检测到的基因数进行过滤(存储在 adata_rna.obs['n_genes_by_counts'])
# nCount (total_counts): 基于每个细胞的总 UMI 数进行过滤(存储在 adata_rna.obs['total_counts'])
# 线粒体基因比例: 基于线粒体转录本占比进行过滤(存储在 adata_rna.obs['pct_counts_mt'])
# 1)按 nFeature 过滤低基因数细胞
sc.pp.filter_cells(adata_rna, min_genes=200) # 等价于按 n_genes_by_counts >= 200 过滤
# 2)过滤低表达基因(在少于 3 个细胞中表达的基因)
sc.pp.filter_genes(adata_rna, min_cells=3)
# 3)按 nCount 和线粒体比例进一步过滤
adata_rna = adata_rna[(adata_rna.obs['n_genes_by_counts'] >= 200) & (adata_rna.obs['n_genes_by_counts'] <= 10000), :].copy() # nFeature 过滤
adata_rna = adata_rna[(adata_rna.obs['total_counts'] >= 200) & (adata_rna.obs['total_counts'] <= 30000), :].copy() # nCount 过滤
adata_rna = adata_rna[adata_rna.obs['pct_counts_mt'] < 20, :].copy() # 线粒体基因比例过滤
print(f"\n质控后细胞数: {adata_rna.n_obs}")
print(f"质控后基因数: {adata_rna.n_vars}")
print(f"过滤掉的细胞数: {n_cells_before - adata_rna.n_obs}")
print(f"过滤掉的基因数: {n_genes_before - adata_rna.n_vars}")
# --- 质控后小提琴图可视化 ---
print("\n=== 质控后质控指标分布 ===")
fig, (ax1, ax2, ax3) = plt.subplots(1, 3, figsize=(10, 4), dpi=80)
sc.pl.violin(adata_rna, keys='n_genes_by_counts', groupby='Sample', rotation=45, ax=ax1, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax1.set_title("Number of Genes", fontsize=10)
ax1.set_ylabel("Gene_Counts", fontsize=9)
ax1.set_xlabel("")
sc.pl.violin(adata_rna, keys='total_counts', groupby='Sample', rotation=45, ax=ax2, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax2.set_title("Total UMI Counts", fontsize=10)
ax2.set_ylabel("UMI_Counts", fontsize=9)
ax2.set_xlabel("")
sc.pl.violin(adata_rna, keys='pct_counts_mt', groupby='Sample', rotation=45, ax=ax3, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax3.set_title("Mitochondrial %", fontsize=10)
ax3.set_ylabel("Percentage(%)", fontsize=9)
ax3.set_xlabel("")
plt.tight_layout()
plt.show()质控前基因数: 38606
=== 质控前质控指标分布 ===
质控后细胞数: 2817
质控后基因数: 19405
过滤掉的细胞数: 11
过滤掉的基因数: 19201
=== 质控后质控指标分布 ===
甲基化数据整合与质控过滤
本节对多个样本的甲基化数据进行整合,并合并质控元数据,然后根据质控指标进行过滤。
数据整合与元数据合并步骤:
- 数据整合:读取各样本的 MCDS 格式甲基化数据并合并。
- 元数据读取:读取每个样本的细胞元数据文件和 Reads 计数文件。
- 甲基化率计算:计算每个细胞的 CpG 和 CH 甲基化率。
- 元数据合并:将计算得到的质控指标合并到
adata_met.obs中。
质控过滤策略:
- total_cpg_number(CpG 位点总数):过滤 CpG 位点总数 < 1000 的细胞,这些细胞可能捕获效率低或质量差。
- CpG%(CpG 甲基化率):过滤 CpG 甲基化率不在 60-100% 范围内的细胞,这些值可能异常。
- CH%(CH 甲基化率):过滤 CH 甲基化率不在 0-5% 范围内的细胞(哺乳动物细胞 CH% 通常 < 5%),异常高值可能提示技术问题。
质控过滤完成后,将统计质控前后细胞数量,并对质控指标进行小提琴图可视化
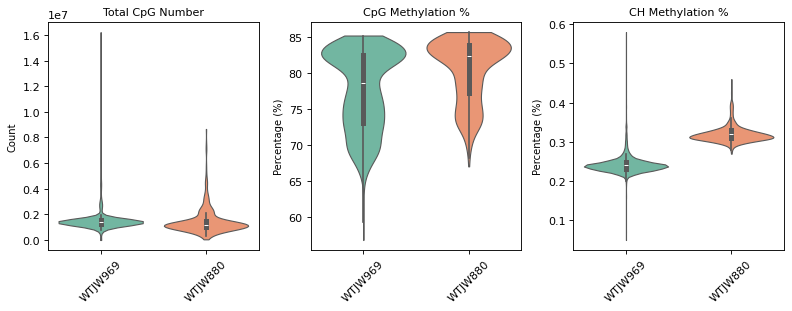
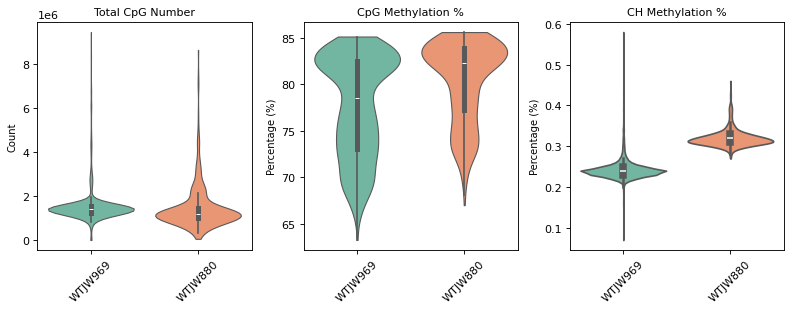
图注(甲基化质控小提琴图):
下图展示了甲基化数据质控过滤前后质控指标的分布情况,按样本 (Sample) 分组显示。
- 质控前小提琴图:显示质控过滤前的质控指标分布,包括
total_cpg_number(CpG 位点总数)、CpG%(CpG 甲基化率)和CH%(CH 甲基化率)。可用于评估原始数据质量和识别异常值。- 质控后小提琴图:显示质控过滤后的质控指标分布。过滤后,低质量细胞(CpG 位点数过少、甲基化率异常)已被去除,数据质量得到提升。
- 横坐标:不同样本 (Sample),便于比较不同样本间的质控指标差异。
- 纵坐标:各质控指标的数值。小提琴图的宽度表示该数值区间的细胞密度,越宽表示该区间的细胞越多。
- 用途:通过对比过滤前后的分布,可以评估质控过滤的效果,确认过滤阈值是否合理。
warnings.filterwarnings('ignore')
# --- 甲基化数据多样本整合 ---
# 读取各样本的 MCDS 格式甲基化数据
obj_met_list = {}
mcds_paths = {}
for i in samples:
# 根据新的文件层级结构构建路径:data/{top_dir}/methylation/{demo_dir}/{sample}/allcools_generate_datasets/{sample}.mcds
top_dir = sample_path_config[i]['top_dir']
demo_dir = sample_path_config[i]['demo_dir']
mcds_path = os.path.join('../../',data_root, top_dir, 'methylation', demo_dir, i, 'allcools_generate_datasets', f'{i}.mcds')
mcds_paths[i] = mcds_path
mcds = MCDS.open(mcds_path, obs_dim='cell', var_dim=var_dim)
adata = mcds.get_score_adata(mc_type=mc_type, quant_type=quant_type, sparse=True)
obj_met_list[i] = adata
# 合并所有样本的数据
adata_met = obj_met_list[samples[0]].concatenate([obj_met_list[i] for i in samples[1:]])
# 保存原始数据
adata_met.layers['raw'] = adata_met.X.copy()
# 添加样本信息
adata_met.obs['Sample'] = adata_met.obs['batch'].map(
{f'{index}': value for index, value in enumerate(samples)}
)
# --- 计算 CpG 和 CH 甲基化率的辅助函数 ---
def compute_cg_ch_level(df):
# 计算 CpG 和 CH 甲基化水平
cg_cov_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_cov')]
cg_mc_col = [i for i in df.columns[df.columns.str.startswith('CG')].to_list() if i.endswith('_mc')]
ch_cov_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_cov')]
ch_mc_col = [i for i in df.columns[df.columns.str.contains('^(CT|CC|CA)')].to_list() if i.endswith('_mc')]
df['CpG_cov'] = df[cg_cov_col].sum(axis=1)
df['CpG_mc'] = df[cg_mc_col].sum(axis=1)
df['CpG%'] = round(df['CpG_mc'] * 100 / df['CpG_cov'], 2)
df['CH_cov'] = df[ch_cov_col].sum(axis=1)
df['CH_mc'] = df[ch_mc_col].sum(axis=1)
df['CH%'] = round(df['CH_mc'] * 100 / df['CH_cov'], 2)
return df
# --- 读取并合并质控元数据 ---
suffix_map = {f'{value}': index for index, value in enumerate(samples)}
meta_list = list()
keep_col = ['barcode', 'total_cpg_number', 'genome_cov', 'reads_counts', 'CpG%', 'CH%']
for i in samples:
# 根据新的文件层级结构构建路径
top_dir = sample_path_config[i]['top_dir']
demo_dir = sample_path_config[i]['demo_dir']
# 读取细胞元数据文件:../../data/{top_dir}/methylation/{demo_dir}/{sample}/split_bams/{sample}_cells.csv
s_cells_meta_path = os.path.join('../../',data_root, top_dir, 'methylation', demo_dir, i, 'split_bams', f'{i}_cells.csv')
s_cells_meta = pd.read_csv(s_cells_meta_path, header=0)
s_cells_meta['barcode'] = [b.replace('_allc.gz', '') for b in s_cells_meta['cell_barcode']]
# 读取 Reads 计数文件:../../data/{top_dir}/methylation/{demo_dir}/{sample}/split_bams/filtered_barcode_reads_counts.csv
s_reads_counts_path = os.path.join('../../',data_root, top_dir, 'methylation', demo_dir, i, 'split_bams', 'filtered_barcode_reads_counts.csv')
s_reads_counts = pd.read_csv(s_reads_counts_path, header=0)
# 合并数据并计算甲基化率
s_merged_df = s_cells_meta.merge(s_reads_counts, how='inner', left_on='barcode', right_on='barcode')
s_merged_df = compute_cg_ch_level(s_merged_df)
s_merged_df = s_merged_df[keep_col]
# 如果是多样本,为 barcode 添加样本标识
if len(samples) > 1:
s_merged_df['barcode'] = [f'{b}-{suffix_map[i]}' for b in s_merged_df['barcode']]
meta_list.append(s_merged_df)
# 合并所有样本的元数据到 adata_met.obs
adata_met.obs = adata_met.obs.merge(
pd.concat(meta_list).set_index('barcode'),
how='left',
left_index=True,
right_index=True
)
# --- 甲基化数据质控过滤 ---
# 保存质控前的统计信息
n_cells_before = adata_met.n_obs
print(f"质控前细胞数: {n_cells_before}")
# --- 质控前小提琴图可视化 ---
print("\n=== 质控前质控指标分布 ===")
fig, (ax1, ax2, ax3) = plt.subplots(1, 3, figsize=(10, 4), dpi=80)
# 1. Total CpG Number
sc.pl.violin(adata_met, keys='total_cpg_number', groupby='Sample', rotation=45, ax=ax1, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax1.set_title("Total CpG Number", fontsize=10)
ax1.set_ylabel("Count", fontsize=9)
ax1.set_xlabel("")
# 2. CpG%
sc.pl.violin(adata_met, keys='CpG%', groupby='Sample', rotation=45, ax=ax2, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax2.set_title("CpG Methylation %", fontsize=10)
ax2.set_ylabel("Percentage (%)", fontsize=9)
ax2.set_xlabel("")
# 3. CH%
sc.pl.violin(adata_met, keys='CH%', groupby='Sample', rotation=45, ax=ax3, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax3.set_title("CH Methylation %", fontsize=10)
ax3.set_ylabel("Percentage (%)", fontsize=9)
ax3.set_xlabel("")
plt.tight_layout()
plt.show()
# 根据质控指标过滤低质量细胞
# total_cpg_number: 过滤 CpG 位点总数过低的细胞(可根据实际情况调整阈值)
# CpG%: 过滤 CpG 甲基化率异常的细胞(通常哺乳动物细胞 CpG% 在 40-80% 之间)
# CH%: 过滤 CH 甲基化率异常的细胞(通常哺乳动物细胞 CH% 较低,< 5%)
# 检查缺失值并处理
if 'total_cpg_number' in adata_met.obs.columns:
adata_met = adata_met[adata_met.obs['total_cpg_number'].notna(), :].copy()
adata_met = adata_met[(adata_met.obs['total_cpg_number'] >= 100) & (adata_met.obs['total_cpg_number'] <= 10000000), :].copy()
if 'CpG%' in adata_met.obs.columns:
adata_met = adata_met[adata_met.obs['CpG%'].notna(), :].copy()
adata_met = adata_met[(adata_met.obs['CpG%'] >= 60) & (adata_met.obs['CpG%'] <= 100), :].copy()
if 'CH%' in adata_met.obs.columns:
adata_met = adata_met[adata_met.obs['CH%'].notna(), :].copy()
adata_met = adata_met[(adata_met.obs['CH%'] >= 0) & (adata_met.obs['CH%'] <= 5), :].copy()
print(f"质控后细胞数: {adata_met.n_obs}")
print(f"过滤掉的细胞数: {n_cells_before - adata_met.n_obs}")
# --- 质控后小提琴图可视化 ---
print("\n=== 质控后质控指标分布 ===")
fig, (ax1, ax2, ax3) = plt.subplots(1, 3, figsize=(10, 4), dpi=80)
# 1. Total CpG Number
sc.pl.violin(adata_met, keys='total_cpg_number', groupby='Sample', rotation=45, ax=ax1, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax1.set_title("Total CpG Number", fontsize=10)
ax1.set_ylabel("Count", fontsize=9)
ax1.set_xlabel("")
# 2. CpG%
sc.pl.violin(adata_met, keys='CpG%', groupby='Sample', rotation=45, ax=ax2, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1, cut=0)
ax2.set_title("CpG Methylation %", fontsize=10)
ax2.set_ylabel("Percentage (%)", fontsize=9)
ax2.set_xlabel("")
# 3. CH%
sc.pl.violin(adata_met, keys='CH%', groupby='Sample', rotation=45, ax=ax3, show=False, palette=my_palette, stripplot=False, size=1, inner='box', linewidth=1.5, cut=0)
ax3.set_title("CH Methylation %", fontsize=10)
ax3.set_ylabel("Percentage (%)", fontsize=9)
ax3.set_xlabel("")
plt.tight_layout()
plt.show()Loading chunk 0-442/442
质控前细胞数: 2638
=== 质控前质控指标分布 ===
质控后细胞数: 2631
过滤掉的细胞数: 7
=== 质控后质控指标分布 ===
RNA 和甲基化数据 barcode 关联、取交集与 doublet 过滤
本节首先根据试剂盒类型关联 RNA 和甲基化数据的 Barcode,然后找出两个数据集中共有的细胞(barcode 交集),最后根据 doublet 表删除 doublet 细胞。
步骤:
- Barcode 关联:根据试剂盒类型(DD-MET3/DD-MET5)关联 RNA 和甲基化数据的 Barcode。
- Barcode 取交集:计算 RNA 和甲基化数据的 barcode 交集,确保两个数据集中的细胞一致。
- Doublet 过滤:读取各样本的 doublet 表,从 RNA 和甲基化数据中删除 doublet 细胞。
根据试剂盒类型关联 RNA 和甲基化数据的 Barcode,建立两个数据集之间的细胞对应关系。
- DD-MET3:RNA 和甲基化数据使用不同的 Barcode,需要通过映射文件关联。请先将 Barcode 映射文件 DD-M_bUCB3_whitelist.csv 下载到本地。 之后读取 Barcode 映射文件(
DD-M_bUCB3_whitelist.csv),建立 RNA Barcode(gex_cb)和甲基化 Barcode(m_cb)之间的对应关系,并创建rna_meta映射表供后续使用。 - DD-MET5:RNA 和甲基化数据使用相同的 Barcode,不需要映射,直接跳过此步骤。
TIP
此步骤建立的映射关系(rna_meta)将用于后续的 doublet 过滤和细胞类型注释转移。
# --- RNA 和甲基化数据关联(支持按样本配置:部分 DD-MET3 需映射、部分 DD-MET5 不映射)---
print("=== 数据状态检查 ===")
print(f"RNA 数据细胞数: {len(adata_rna.obs)}")
print(f"甲基化数据细胞数: {len(adata_met.obs)}")
if len(adata_rna.obs) == 0:
raise ValueError("RNA 数据为空,无法建立映射关系。请检查前面的数据加载和质控步骤。")
if len(adata_met.obs) == 0:
raise ValueError("甲基化数据为空,无法建立映射关系。请检查前面的质控过滤步骤。")
suffix_map = {s: i for i, s in enumerate(samples)} # sample -> batch index
# 若存在 DD-MET3 样本,则加载 barcode 映射文件(只加载一次)
if any(reagent_type_map.get(s, reagent_type) == "DD-MET3" for s in samples):
mapping_file = "DD-M_bUCB3_whitelist.csv"
if not os.path.exists(mapping_file):
raise FileNotFoundError("未找到映射文件:DD-M_bUCB3_whitelist.csv,请确认文件路径。")
gex_mc_bc_map = pd.read_csv(mapping_file, index_col=None)
if 'gex_cb' not in gex_mc_bc_map.columns or 'm_cb' not in gex_mc_bc_map.columns:
raise ValueError(f"映射文件格式错误:应包含 'gex_cb' 和 'm_cb' 列,实际列名: {list(gex_mc_bc_map.columns)}")
print(f"映射文件路径: {mapping_file},记录数: {len(gex_mc_bc_map)}")
else:
gex_mc_bc_map = None
rna_meta_list = []
for sample in samples:
rt = reagent_type_map.get(sample, reagent_type)
rna_obs_s = adata_rna.obs[adata_rna.obs['Sample'] == sample]
met_barcodes_s = set(adata_met.obs.index[adata_met.obs['Sample'] == sample])
suf = suffix_map[sample]
if rt == "DD-MET3":
# 该样本需要 barcode 映射
rm = rna_obs_s.copy()
rm["rna_bc_with_suffix"] = rm.index
rm["gex_cb"] = [re.sub(r'-.*$', '', b) for b in rm["rna_bc_with_suffix"]]
rm = rm.merge(gex_mc_bc_map, how='left', left_on='gex_cb', right_on='gex_cb')
rm = rm.dropna(subset=['m_cb'])
rm["m_cb"] = rm["m_cb"].astype(str) + "-" + str(suf)
rm = rm[rm["m_cb"].isin(met_barcodes_s)]
rm.index = rm["m_cb"]
rna_meta_list.append(rm)
print(f" {sample} (DD-MET3): 映射后保留 {len(rm)} 个共同细胞")
else:
# DD-MET5:同一 barcode,只保留 RNA 与甲基化都有的细胞
common_s = set(rna_obs_s.index) & met_barcodes_s
rm = pd.DataFrame({"rna_bc_with_suffix": list(common_s), "m_cb": list(common_s), "gex_cb": [re.sub(r'-.*$', '', b) for b in common_s]})
rm.index = rm["m_cb"]
rna_meta_list.append(rm)
print(f" {sample} (DD-MET5): 不映射,共有 {len(rm)} 个共同细胞")
if rna_meta_list:
rna_meta = pd.concat(rna_meta_list, axis=0)
rna_met_mapping = rna_meta[["gex_cb", "m_cb", "rna_bc_with_suffix"]].copy()
print(f"\n=== 汇总 ===")
print(f"成功建立关联的细胞数: {len(rna_meta)}")
else:
rna_meta = None
rna_met_mapping = NoneRNA 数据细胞数: 2817
甲基化数据细胞数: 2631
映射文件路径: DD-M_bUCB3_whitelist.csv,记录数: 248832
WTJW969 (DD-MET5): 不映射,共有 2187 个共同细胞
WTJW880 (DD-MET3): 映射后保留 438 个共同细胞
=== 汇总 ===
成功建立关联的细胞数: 2625
# --- RNA 和甲基化数据 barcode 取交集并过滤 ---
# 由于 RNA 和甲基化数据分别进行了质控过滤,需要确保两个数据集中的细胞一致
# 若存在映射表 rna_meta(含 DD-MET3 或混合样本),则按映射取交集;否则按 barcode 直接取交集(全为 DD-MET5)
print("=== RNA 和甲基化数据 barcode 取交集 ===")
if rna_meta is not None and len(rna_meta) > 0:
# DD-MET3:通过映射关系取交集
# rna_meta 的索引是甲基化 barcode(m_cb),包含了对应的 RNA barcode 信息
# 使用 rna_meta 中已有的映射关系来取交集
if rna_meta is None or len(rna_meta) == 0:
raise ValueError("rna_meta 为空,请先执行 barcode 关联步骤(Cell 15)")
# 获取映射表中有效的甲基化 barcode(这些是已经建立映射关系的)
mapped_met_barcodes = set(rna_meta.index)
# 获取甲基化数据中实际存在的 barcode
met_barcodes = set(adata_met.obs.index)
# 获取 RNA 数据中实际存在的 barcode
rna_barcodes_with_suffix = set(adata_rna.obs.index)
print(f"RNA 数据细胞数: {len(rna_barcodes_with_suffix)}")
print(f"甲基化数据细胞数: {len(met_barcodes)}")
print(f"映射表中有效的甲基化 barcode 数: {len(mapped_met_barcodes)}")
# 取交集:映射表中存在的且甲基化数据中也存在的 barcode
common_met_barcodes = mapped_met_barcodes & met_barcodes
print(f"映射表与甲基化数据的交集(甲基化 barcode): {len(common_met_barcodes)}")
if len(common_met_barcodes) == 0:
print("⚠️ 警告:映射表与甲基化数据没有交集!")
print("可能的原因:")
print("1. 映射关系建立失败")
print("2. 甲基化数据在质控步骤中被过度过滤")
print("3. barcode 格式不匹配")
raise ValueError("无法找到交集,请检查映射关系和数据状态。")
# 通过映射关系,找到对应的 RNA barcode(带后缀)
rna_meta_filtered = rna_meta.loc[list(common_met_barcodes), :]
common_rna_barcodes_with_suffix = set(rna_meta_filtered["rna_bc_with_suffix"].values)
print(f"通过映射关系找到的 RNA barcode 数: {len(common_rna_barcodes_with_suffix)}")
# 确保 RNA barcode 在 RNA 数据中存在
common_rna_barcodes_with_suffix = common_rna_barcodes_with_suffix & rna_barcodes_with_suffix
print(f"RNA barcode 与 RNA 数据的交集: {len(common_rna_barcodes_with_suffix)}")
if len(common_rna_barcodes_with_suffix) == 0:
print("⚠️ 警告:映射后的 RNA barcode 在 RNA 数据中不存在!")
raise ValueError("无法找到有效的 RNA barcode,请检查映射关系和 RNA 数据。")
# 更新对应的甲基化 barcode(只保留 RNA barcode 也存在的)
rna_meta_filtered = rna_meta_filtered[rna_meta_filtered["rna_bc_with_suffix"].isin(common_rna_barcodes_with_suffix)]
common_met_barcodes = set(rna_meta_filtered.index)
print(f"\n最终交集统计:")
print(f" 交集细胞数(甲基化 barcode): {len(common_met_barcodes)}")
print(f" 交集细胞数(RNA barcode): {len(common_rna_barcodes_with_suffix)}")
# 对两个数据集都进行过滤,只保留交集部分的细胞
print(f"\n开始过滤数据...")
adata_rna_before = adata_rna.n_obs
adata_met_before = adata_met.n_obs
adata_rna = adata_rna[adata_rna.obs.index.isin(common_rna_barcodes_with_suffix), :].copy()
adata_met = adata_met[adata_met.obs.index.isin(common_met_barcodes), :].copy()
# 更新 rna_meta,只保留过滤后的映射关系
rna_meta = rna_meta.loc[list(common_met_barcodes), :]
print(f"\n过滤结果:")
print(f" RNA 数据: {adata_rna_before} → {adata_rna.n_obs} (删除 {adata_rna_before - adata_rna.n_obs} 个)")
print(f" 甲基化数据: {adata_met_before} → {adata_met.n_obs} (删除 {adata_met_before - adata_met.n_obs} 个)")
# 验证过滤后的一致性
if adata_rna.n_obs != adata_met.n_obs:
print(f"⚠️ 警告:过滤后 RNA 和甲基化数据的细胞数不一致!")
print(f" RNA: {adata_rna.n_obs}, 甲基化: {adata_met.n_obs}")
else:
print(f"✅ 过滤后两个数据集的细胞数一致: {adata_rna.n_obs}")
else:
# 无映射表或为空:RNA 和甲基化使用相同 Barcode,直接取交集(全为 DD-MET5 且无细胞时也会进此分支)
rna_barcodes = set(adata_rna.obs.index)
met_barcodes = set(adata_met.obs.index)
# 计算交集
common_barcodes = rna_barcodes & met_barcodes
print(f"RNA 数据细胞数: {len(rna_barcodes)}")
print(f"甲基化数据细胞数: {len(met_barcodes)}")
print(f"交集细胞数: {len(common_barcodes)}")
print(f"仅在 RNA 数据中的细胞数: {len(rna_barcodes - met_barcodes)}")
print(f"仅在甲基化数据中的细胞数: {len(met_barcodes - rna_barcodes)}")
if len(common_barcodes) == 0:
print("⚠️ 警告:RNA 和甲基化数据没有交集!")
print("可能的原因:")
print("1. 两个数据集的 barcode 格式不一致")
print("2. 质控过滤后没有共同细胞")
raise ValueError("无法找到交集,请检查数据状态和 barcode 格式。")
# 对两个数据集都进行过滤,只保留交集部分的细胞
print(f"\n开始过滤数据...")
adata_rna_before = adata_rna.n_obs
adata_met_before = adata_met.n_obs
adata_rna = adata_rna[adata_rna.obs.index.isin(common_barcodes), :].copy()
adata_met = adata_met[adata_met.obs.index.isin(common_barcodes), :].copy()
print(f"\n过滤结果:")
print(f" RNA 数据: {adata_rna_before} → {adata_rna.n_obs} (删除 {adata_rna_before - adata_rna.n_obs} 个)")
print(f" 甲基化数据: {adata_met_before} → {adata_met.n_obs} (删除 {adata_met_before - adata_met.n_obs} 个)")
# 验证过滤后的一致性
if adata_rna.n_obs != adata_met.n_obs:
print(f"⚠️ 警告:过滤后 RNA 和甲基化数据的细胞数不一致!")
print(f" RNA: {adata_rna.n_obs}, 甲基化: {adata_met.n_obs}")
else:
print(f"✅ 过滤后两个数据集的细胞数一致: {adata_rna.n_obs}")RNA 数据细胞数: 2817
甲基化数据细胞数: 2631
映射表中有效的甲基化 barcode 数: 2625
映射表与甲基化数据的交集(甲基化 barcode): 2625
通过映射关系找到的 RNA barcode 数: 2625
RNA barcode 与 RNA 数据的交集: 2625
最终交集统计:
交集细胞数(甲基化 barcode): 2625
交集细胞数(RNA barcode): 2625
开始过滤数据...n
过滤结果:
RNA 数据: 2817 → 2625 (删除 192 个)
甲基化数据: 2631 → 2625 (删除 6 个)
✅ 过滤后两个数据集的细胞数一致: 2625
根据 doublet 表删除双细胞
本节根据各样本的 doublet 识别结果,从 RNA 和甲基化数据中删除双细胞 (doublet)。
数据来源:{sample}_doublet.txt 文件来自 甲基化+RNA 双组学-双细胞判断。ipynb 的分析结果,包含每个样本中识别出的双细胞信息。
处理逻辑:
- 读取各样本的 doublet 表(
{sample}_doublet.txt),获取甲基化数据的 doublet barcode(m_cb列)。 - 根据试剂盒类型进行不同的处理:
- DD-MET3:通过映射文件将甲基化 barcode 转换为 RNA barcode,然后分别从两个数据集中删除。
- DD-MET5:甲基化 barcode 就是 RNA barcode,直接使用相同的 barcode 从两个数据集中删除。
- 输出删除前后的细胞数统计信息。
TIP
如果不需要去除双细胞,可以跳过此单元格。
# --- 根据 doublet 表删除 doublet ---
doublet_df_column_name = "met_is_doublet"
# 读取各样本的 doublet 表并合并
doublet_met_barcodes = set() # 甲基化数据的 doublet barcode(用于删除甲基化数据)
doublet_rna_barcodes = set() # RNA 数据的 doublet barcode(用于删除 RNA 数据)
suffix_map = {f'{value}': index for index, value in enumerate(samples)}
for sample in samples:
doublet_path = f"{sample}_doublet.txt"
if os.path.exists(doublet_path):
doublet_df = pd.read_csv(doublet_path, sep='\t')
# 根据 doublet_df 的列名来决定如何识别双细胞
if doublet_df_column_name == 'met_is_doublet':
# 如果列名是 "met_is_doublet",使用 met_is_doublet == True 来识别双细胞
sample_doublets_met = doublet_df[doublet_df['met_is_doublet'] == True]['m_cb'].tolist()
elif doublet_df_column_name == 'cell_multi_highlight':
# 如果列名是 "cell_multi_highlight",则列名下的 'multi_cells' 是双细胞
sample_doublets_met = doublet_df[doublet_df['cell_multi_highlight'] == 'multi_cells']['m_cb'].tolist()
elif doublet_df_column_name == 'cell_multi_highlight':
sample_doublets_met = doublet_df[doublet_df['rna_doublet'] == 'doublet']['m_cb'].tolist()
else:
raise ValueError(f"doublet 文件 {doublet_path} 中未找到预期的列名。期望列名:'met_is_doublet' 或 'cell_multi_highlight',实际列名:{list(doublet_df.columns)}")
# 为甲基化 barcode 添加样本后缀(用于从甲基化数据中删除)
if len(samples) > 1:
sample_doublets_met_with_suffix = [f"{b}-{suffix_map[sample]}" for b in sample_doublets_met]
else:
sample_doublets_met_with_suffix = sample_doublets_met
doublet_met_barcodes.update(sample_doublets_met_with_suffix)
rt = reagent_type_map.get(sample, reagent_type)
# 根据试剂盒类型进行不同的处理
if rt == "DD-MET3":
# DD-MET3:需要将甲基化 barcode 转换为 RNA barcode
# 通过映射文件将甲基化 barcode 转换为 RNA barcode
sample_gex_mc_map = gex_mc_bc_map[gex_mc_bc_map['m_cb'].isin(sample_doublets_met)]
sample_doublets_rna = sample_gex_mc_map['gex_cb'].tolist()
# 为 RNA barcode 添加样本后缀
if len(samples) > 1:
sample_doublets_rna_with_suffix = [f"{b}-{suffix_map[sample]}" for b in sample_doublets_rna]
else:
sample_doublets_rna_with_suffix = sample_doublets_rna
doublet_rna_barcodes.update(sample_doublets_rna_with_suffix)
elif rt == "DD-MET5":
# DD-MET5:甲基化 barcode 就是 RNA barcode,直接使用
doublet_rna_barcodes.update(sample_doublets_met_with_suffix)
else:
raise ValueError(f"不支持的试剂盒类型: {reagent_type},请设置为 'DD-MET3' 或 'DD-MET5'")
print(f"{sample}: 检测到 {len(sample_doublets_met)} 个 doublet 细胞")
else:
print(f"警告:未找到 {sample} 的 doublet 文件 {doublet_path}")
if doublet_met_barcodes:
print(f"\n总共检测到的 doublet 细胞数: {len(doublet_met_barcodes)}")
print(f"试剂盒类型: {reagent_type}")
# 从甲基化数据中删除 doublet(使用甲基化 barcode)
adata_met = adata_met[~adata_met.obs.index.isin(doublet_met_barcodes), :].copy()
# 从 RNA 数据中删除 doublet(使用 RNA barcode)
adata_rna = adata_rna[~adata_rna.obs.index.isin(doublet_rna_barcodes), :].copy()
print(f"删除 doublet 后 RNA 数据细胞数: {adata_rna.n_obs}")
print(f"删除 doublet 后甲基化数据细胞数: {adata_met.n_obs}")
else:
print("警告:未找到任何 doublet 文件,跳过 doublet 过滤")WTJW880: 检测到 134 个 doublet 细胞
总共检测到的 doublet 细胞数: 406
试剂盒类型: DD-MET3
删除 doublet 后 RNA 数据细胞数: 2226
删除 doublet 后甲基化数据细胞数: 2226
单细胞 RNA 测序数据多样本整合分析流程
本节对质控后的 RNA 数据进行标准化、特征选择、降维、批次校正和聚类分析。
数据标准化与降维分析
warnings.filterwarnings('ignore')
# --- RNA 数据标准化与降维分析 ---
# 标准化:总计数归一化,消除细胞间测序深度差异
sc.pp.normalize_total(adata_rna, inplace=True)
# Log 转换:log(x + 1)
sc.pp.log1p(adata_rna)
# 识别高变基因(2000个),这些基因最能反映细胞间的生物学差异
sc.pp.highly_variable_genes(adata_rna, n_bins=100, n_top_genes=2000)
# 保存原始数据,然后仅保留高变基因
adata_rna.raw = adata_rna
adata_rna = adata_rna[:, adata_rna.var.highly_variable]
# 高变基因 Z-score 标准化
sc.pp.scale(adata_rna, max_value=10)
# 主成分分析(PCA)
sc.tl.pca(adata_rna)
# Harmony 批次效应校正
ho = run_harmony(
adata_rna.obsm['X_pca'],
meta_data=adata_rna.obs,
vars_use='Sample',
random_state=0,
max_iter_harmony=20
)
adata_rna.obsm['X_pca_harmony'] = ho.Z_corr.T
# 使用 Harmony 校正后的结果进行后续分析
reduc_use = 'X_pca_harmony'
# 构建 KNN 图
sc.pp.neighbors(adata_rna, n_neighbors=30, use_rep=reduc_use)
# UMAP 降维可视化
sc.tl.umap(adata_rna)
# t-SNE 降维可视化
sc.tl.tsne(adata_rna, use_rep=reduc_use)
# Leiden 聚类
sc.tl.leiden(adata_rna, resolution=1)2026-01-28 16:12:38,318 - harmonypy - INFO - Iteration 1 of 20
2026-01-28 16:12:40,373 - harmonypy - INFO - Iteration 2 of 20
2026-01-28 16:12:42,297 - harmonypy - INFO - Converged after 2 iterations
RNA 单组细胞注释
图注(RNA UMAP - Leiden 聚类):
下图展示了 RNA 数据在 UMAP 降维空间中的细胞分布,按 Leiden 聚类结果着色。
- 横坐标和纵坐标:UMAP 降维后的两个主维度(UMAP1 和 UMAP2),用于在二维空间中展示高维数据的细胞相似性关系。
- 颜色:不同颜色代表不同的 Leiden 聚类结果,每个聚类可能对应一种或多种细胞类型。
- 点:每个点代表一个细胞,位置反映该细胞与其他细胞的相似性。相似细胞在 UMAP 空间中聚集在一起。
- 用途:用于评估聚类效果,识别细胞亚群,以及发现潜在的细胞类型。
warnings.filterwarnings('ignore')
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(6, 6))
sc.pl.umap(adata_rna, color='leiden', s=35, palette=my_palette, frameon=True)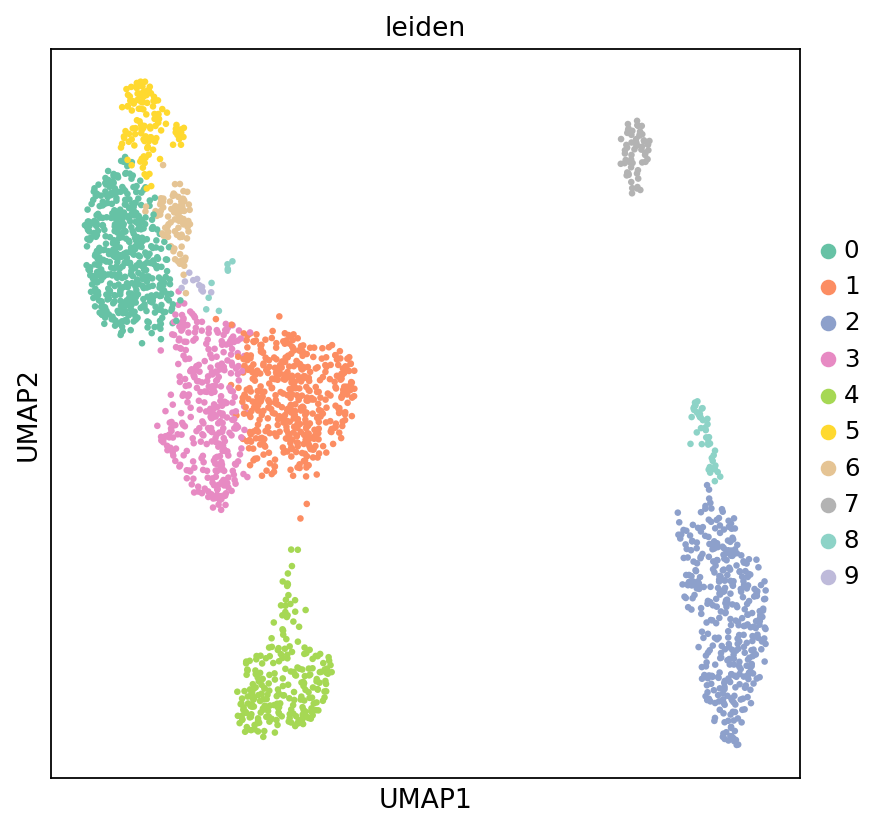
图注(RNA UMAP - 标志基因表达):
下图展示了 RNA 数据在 UMAP 降维空间中标志基因的表达分布。
- 横坐标和纵坐标:UMAP 降维后的两个主维度(UMAP1 和 UMAP2)。
- 颜色:颜色深浅表示各标志基因的表达水平,颜色越深表示表达量越高。展示的标志基因包括:
CD3D(T 细胞)、NKG7(NK 细胞)、CD8A(CD8+ T 细胞)、CD4(CD4+ T 细胞)、CD79A和MS4A1(B 细胞)、JCHAIN(浆细胞)、LYZ(单核细胞)、LILRA4(pDC 细胞)。- 点:每个点代表一个细胞。
- 用途:通过标志基因的表达模式,可以初步识别和验证不同细胞类型,辅助细胞类型注释。
colors = ["#F5E6CC", "#FDBB84", "#E34A33", "#7F0000"]
my_warm_cmap = mcolors.LinearSegmentedColormap.from_list("white_orange_red", colors, N=256)
marker_names = ["CD3D","NKG7","CD8A","CD4","CCR7","CD79A","MS4A1","LYZ","FCN1","FCGR3A","CST3"]
sc.pl.umap(
adata_rna,
color=marker_names,
ncols=4,
color_map=my_warm_cmap,
vmax='p99',
s=10,
frameon=False,
vmin=0,
show=False
)
plt.gcf().set_size_inches(16, 8)
plt.show()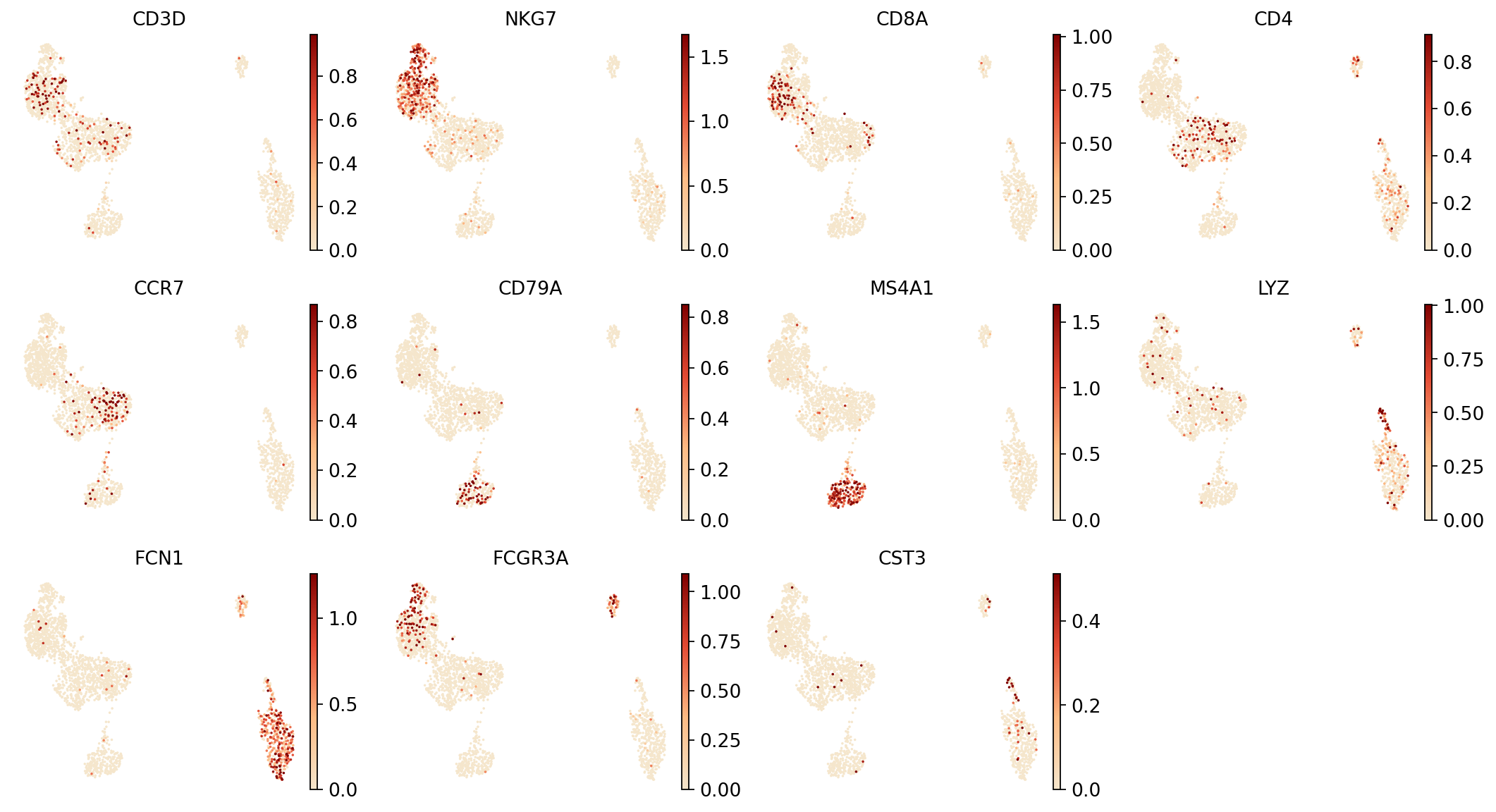
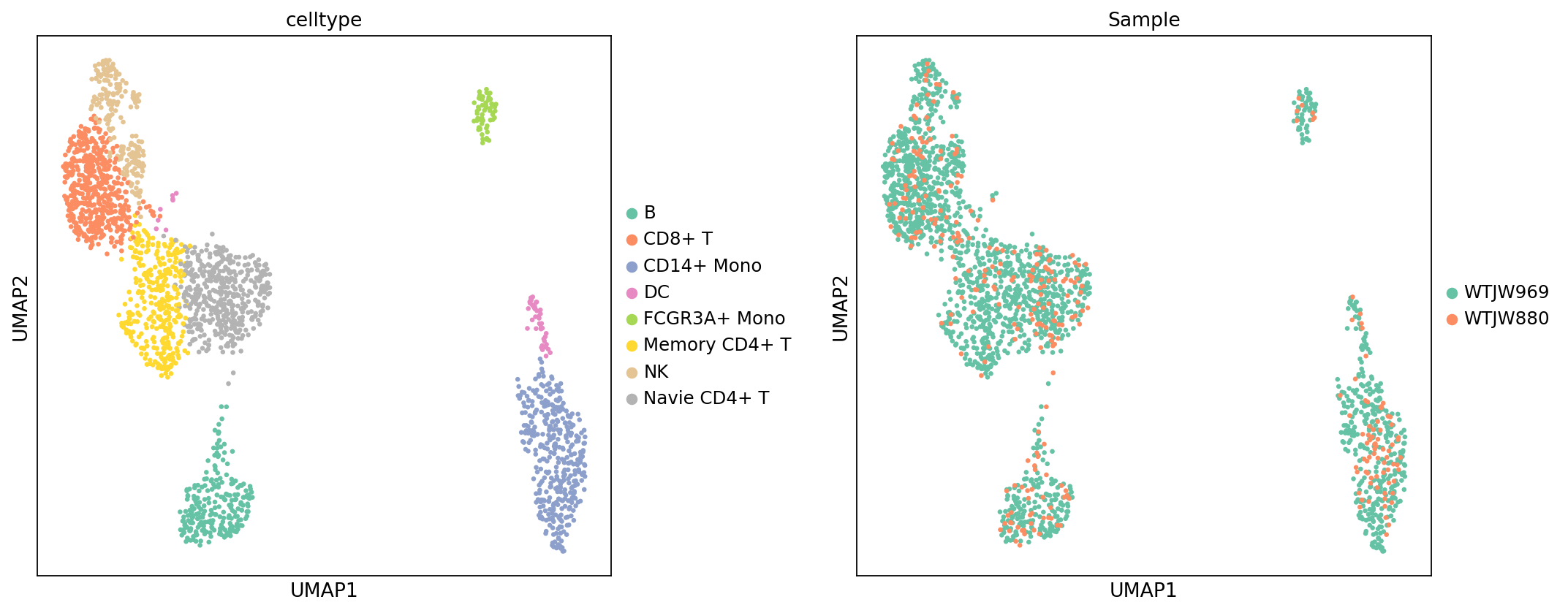
图注(RNA UMAP - 细胞类型和样本):
下图展示了 RNA 数据在 UMAP 降维空间中的分布,按细胞类型 (celltype) 和样本 (Sample) 着色。
- 横坐标和纵坐标:UMAP 降维后的两个主维度(UMAP1 和 UMAP2)。
- 颜色(celltype 图):不同颜色代表不同的细胞类型,用于展示细胞类型注释后的整体分布情况。
- 颜色(Sample 图):不同颜色代表不同的样本,用于评估样本间的批次效应及细胞类型组成差异。
- 点:每个点代表一个细胞。
- 用途:评估细胞类型注释效果,检查样本间的批次效应,以及比较不同样本的细胞类型组成。
celltype = {
'0': 'CD8+ T',
'1': 'Navie CD4+ T',
'2': 'CD14+ Mono',
'3': 'Memory CD4+ T',
'4': 'B',
'5': 'NK',
'6': 'NK',
'7': 'FCGR3A+ Mono',
'8': 'DC',
'9': 'CD8+ T'
}
adata_rna.obs['celltype'] = adata_rna.obs['leiden'].map(celltype)sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(6, 6))
sc.pl.umap(
adata_rna,
color=['celltype', 'Sample'],
ncols=2,
s=35,
wspace=0.3,
palette=my_palette
)
adata_rna.write("adata_rna.h5ad")单细胞甲基化测序数据多样本整合分析流程
本节对质控后的甲基化数据进行预处理、降维、批次校正、聚类分析和细胞类型注释转移。
数据预处理与降维分析
本节对质控后的甲基化数据进行预处理、降维聚类和批次校正。
# --- 甲基化数据预处理与降维分析 ---
# 矩阵二值化:设置 95% 阈值进行数据二值化,突出显著的表观遗传差异
binarize_matrix(adata_met, cutoff=0.95)
# 区域过滤:智能过滤基因组区域
filter_regions(adata_met)
# LSI 降维:采用 ARPACK 算法进行线性降维,提取基因组甲基化模式的主成分
lsi(adata_met, algorithm='arpack', obsm='X_lsi')
# 显著性主成分检验:仅保留 p < 0.1 的显著主成分
significant_pc_test(adata_met, p_cutoff=0.1, obsm='X_lsi', update=True)
# Harmony 批次效应校正
ho = run_harmony(
adata_met.obsm['X_lsi'],
meta_data=adata_met.obs,
vars_use='Sample',
random_state=0,
max_iter_harmony=20
)
adata_met.obsm['X_lsi_harmony'] = ho.Z_corr.T
# 使用 Harmony 校正后的结果进行后续分析
reduc_use = 'X_lsi_harmony'
# 构建 KNN 图
sc.pp.neighbors(adata_met, n_neighbors=30, use_rep=reduc_use)
# UMAP 降维可视化
sc.tl.umap(adata_met)
# t-SNE 降维可视化
sc.tl.tsne(adata_met, use_rep=reduc_use)
# Leiden 聚类
sc.tl.leiden(adata_met, resolution=0.5)2026-01-28 16:13:44,567 - harmonypy - INFO - Computing initial centroids with sklearn.KMeans...n
13 components passed P cutoff of 0.1.
Changing adata.obsm['X_pca'] from shape (2226, 100) to (2226, 13)
2026-01-28 16:13:44,833 - harmonypy - INFO - sklearn.KMeans initialization complete.
2026-01-28 16:13:44,950 - harmonypy - INFO - Iteration 1 of 20
2026-01-28 16:13:49,462 - harmonypy - INFO - Iteration 2 of 20
2026-01-28 16:13:49,850 - harmonypy - INFO - Iteration 3 of 20
2026-01-28 16:13:52,530 - harmonypy - INFO - Iteration 4 of 20
2026-01-28 16:13:52,999 - harmonypy - INFO - Iteration 5 of 20
2026-01-28 16:13:53,363 - harmonypy - INFO - Iteration 6 of 20
2026-01-28 16:13:54,649 - harmonypy - INFO - Iteration 7 of 20
2026-01-28 16:13:56,828 - harmonypy - INFO - Converged after 7 iterations
11 components passed P cutoff of 0.1.
Changing adata.obsm['X_pca'] from shape (6307, 100) to (6307, 11)
2026-01-12 17:33:48,052 - harmonypy - INFO - sklearn.KMeans initialization complete.
2026-01-12 17:33:48,177 - harmonypy - INFO - Iteration 1 of 20
2026-01-12 17:33:55,923 - harmonypy - INFO - Iteration 2 of 20
2026-01-12 17:34:04,649 - harmonypy - INFO - Iteration 3 of 20
2026-01-12 17:34:12,981 - harmonypy - INFO - Iteration 4 of 20
2026-01-12 17:34:22,877 - harmonypy - INFO - Iteration 5 of 20
2026-01-12 17:34:32,578 - harmonypy - INFO - Iteration 6 of 20
2026-01-12 17:34:41,337 - harmonypy - INFO - Iteration 7 of 20
2026-01-12 17:34:47,408 - harmonypy - INFO - Iteration 8 of 20
2026-01-12 17:34:52,370 - harmonypy - INFO - Iteration 9 of 20
2026-01-12 17:34:57,463 - harmonypy - INFO - Iteration 10 of 20
2026-01-12 17:35:01,333 - harmonypy - INFO - Iteration 11 of 20
2026-01-12 17:35:04,390 - harmonypy - INFO - Iteration 12 of 20
2026-01-12 17:35:08,876 - harmonypy - INFO - Iteration 13 of 20
2026-01-12 17:35:13,944 - harmonypy - INFO - Iteration 14 of 20
2026-01-12 17:35:19,234 - harmonypy - INFO - Converged after 14 iterations
细胞类型注释转移
本节将 RNA 数据的细胞类型注释转移到甲基化数据,实现多组学数据的细胞类型统一标注。
注释转移策略:
根据试剂盒类型(reagent_type)采用不同的转移方法:
DD-MET3 试剂盒:
- RNA 和甲基化数据使用不同的 Barcode,需要通过之前建立的映射关系(
rna_meta)进行转移。 - 通过
rna_bc_with_suffix(RNA barcode,带后缀)从adata_rna.obs中获取celltype。 - 然后将
celltype映射到甲基化数据的m_cb(甲基化 barcode)。 - 仅对 RNA 与甲基化数据集中同时存在的细胞进行注释转移。
- RNA 和甲基化数据使用不同的 Barcode,需要通过之前建立的映射关系(
DD-MET5 试剂盒:
- RNA 和甲基化数据使用相同的 Barcode,可以直接按索引对齐转移。
- 计算两个数据集的 barcode 交集,仅对两个数据集中共有的细胞进行注释转移。
WARNING
执行此步骤前,请确保:
- RNA 数据已完成细胞类型注释(
adata_rna.obs中存在celltype列)。 - 对于 DD-MET3 试剂盒,已执行 barcode 关联步骤和取交集步骤。
# --- 将 RNA 数据的细胞类型注释转移到甲基化数据 ---
print("=== 细胞类型注释转移 ===")
# 检查 RNA 数据是否有 celltype 列
if 'celltype' not in adata_rna.obs.columns:
raise ValueError("RNA 数据中没有 'celltype' 列,请先对 RNA 数据进行细胞类型注释。")
print(f"RNA 数据中的细胞类型数: {adata_rna.obs['celltype'].nunique()}")
print(f"RNA 数据中的细胞类型: {sorted(adata_rna.obs['celltype'].unique())}")
# 根据试剂盒类型进行不同的处理
if rna_meta is not None and len(rna_meta) > 0:
#if reagent_type == "DD-MET3":
# DD-MET3:通过之前建立的映射关系转移细胞类型注释
# rna_meta 的索引是 m_cb(甲基化 barcode),包含 rna_bc_with_suffix(RNA barcode,带后缀)
if 'rna_meta' not in locals() or rna_meta is None:
raise ValueError("rna_meta 不存在,请先执行 barcode 关联步骤(Cell 15)和取交集步骤(Cell 16)。")
if len(rna_meta) == 0:
raise ValueError("rna_meta 为空,无法进行注释转移。请检查映射关系。")
# 从当前的 adata_rna.obs 中获取 celltype(因为 rna_meta 可能是在 celltype 注释之前创建的)
# 通过 rna_bc_with_suffix 从 adata_rna.obs 中获取 celltype
print(f"\n通过映射关系转移注释...")
print(f"rna_meta 中的映射关系数: {len(rna_meta)}")
print(f"甲基化数据中的细胞数: {len(adata_met.obs)}")
# 通过 rna_meta 的 rna_bc_with_suffix 获取对应的 celltype
# rna_meta 的索引是 m_cb(甲基化 barcode),包含 rna_bc_with_suffix(RNA barcode,带后缀)
# 步骤:
# 1. 从 rna_meta 中提取 rna_bc_with_suffix 和对应的 m_cb(索引)
# 2. 从 adata_rna.obs 中获取 celltype(通过 rna_bc_with_suffix)
# 3. 将 celltype 映射到甲基化数据的 m_cb
# 创建从 RNA barcode 到 celltype 的映射
rna_celltype_map = adata_rna.obs['celltype'].to_dict()
# 从 rna_meta 中获取 rna_bc_with_suffix,并映射到 celltype
# 只保留在 adata_met.obs.index 中的 barcode
met_barcodes_in_meta = rna_meta.index.intersection(adata_met.obs.index)
rna_meta_filtered = rna_meta.loc[list(met_barcodes_in_meta), :]
# 通过 rna_bc_with_suffix 从 adata_rna.obs 中获取 celltype
celltype_series = rna_meta_filtered['rna_bc_with_suffix'].map(rna_celltype_map)
# 检查是否有缺失值
missing_in_map = celltype_series.isna().sum()
if missing_in_map > 0:
missing_barcodes = celltype_series[celltype_series.isna()].index
print(f"⚠️ 警告:有 {missing_in_map} 个细胞的 RNA barcode 在 adata_rna.obs 中找不到对应的 celltype")
print(f" 示例 barcode: {list(missing_barcodes[:5])}")
# 将 celltype 赋值给甲基化数据(按索引对齐)
adata_met.obs["celltype"] = celltype_series
# 检查是否有缺失值
missing_count = adata_met.obs["celltype"].isna().sum()
if missing_count > 0:
print(f"⚠️ 警告:有 {missing_count} 个细胞无法找到对应的 celltype 注释")
# 可以选择删除这些细胞或填充为 "Unknown"
# adata_met.obs["celltype"] = adata_met.obs["celltype"].fillna("Unknown")
else:
print(f"✅ 成功转移 {len(adata_met.obs)} 个细胞的 celltype 注释")
adata_met.obs["celltype"] = adata_met.obs["celltype"].astype('category')
print(f"\n转移后的细胞类型统计:")
print(adata_met.obs['celltype'].value_counts())
else:
#elif reagent_type == "DD-MET5":
# DD-MET5:RNA 和甲基化数据使用相同的 Barcode,直接转移细胞类型注释
# 由于 barcode 相同,直接按索引对齐转移细胞类型注释
# 只转移两个数据集中都存在的细胞
print(f"\n直接按索引对齐转移注释...")
print(f"RNA 数据细胞数: {len(adata_rna.obs)}")
print(f"甲基化数据细胞数: {len(adata_met.obs)}")
# 计算交集
common_barcodes_for_annotation = set(adata_rna.obs.index) & set(adata_met.obs.index)
print(f"共有的 barcode 数: {len(common_barcodes_for_annotation)}")
if len(common_barcodes_for_annotation) == 0:
raise ValueError("RNA 和甲基化数据没有共有的 barcode,无法进行注释转移。")
# 直接按索引对齐转移
adata_met.obs["celltype"] = adata_rna.obs.loc[list(common_barcodes_for_annotation), "celltype"]
# 检查是否有缺失值
missing_count = adata_met.obs["celltype"].isna().sum()
if missing_count > 0:
print(f"⚠️ 警告:有 {missing_count} 个细胞无法找到对应的 celltype 注释")
else:
print(f"✅ 成功转移 {len(common_barcodes_for_annotation)} 个细胞的 celltype 注释")
adata_met.obs["celltype"] = adata_met.obs["celltype"].astype('category')
print(f"\n转移后的细胞类型统计:")
print(adata_met.obs['celltype'].value_counts())RNA 数据中的细胞类型数: 8
RNA 数据中的细胞类型: ['B', 'CD14+ Mono', 'CD8+ T', 'DC', 'FCGR3A+ Mono', 'Memory CD4+ T', 'NK', 'Navie CD4+ T']
通过映射关系转移注释...n rna_meta 中的映射关系数: 2625
甲基化数据中的细胞数: 2226
✅ 成功转移 2226 个细胞的 celltype 注释
转移后的细胞类型统计:
celltype
CD8+ T 499
Navie CD4+ T 456
CD14+ Mono 379
Memory CD4+ T 338
B 240
NK 200
FCGR3A+ Mono 62
DC 52
Name: count, dtype: int64
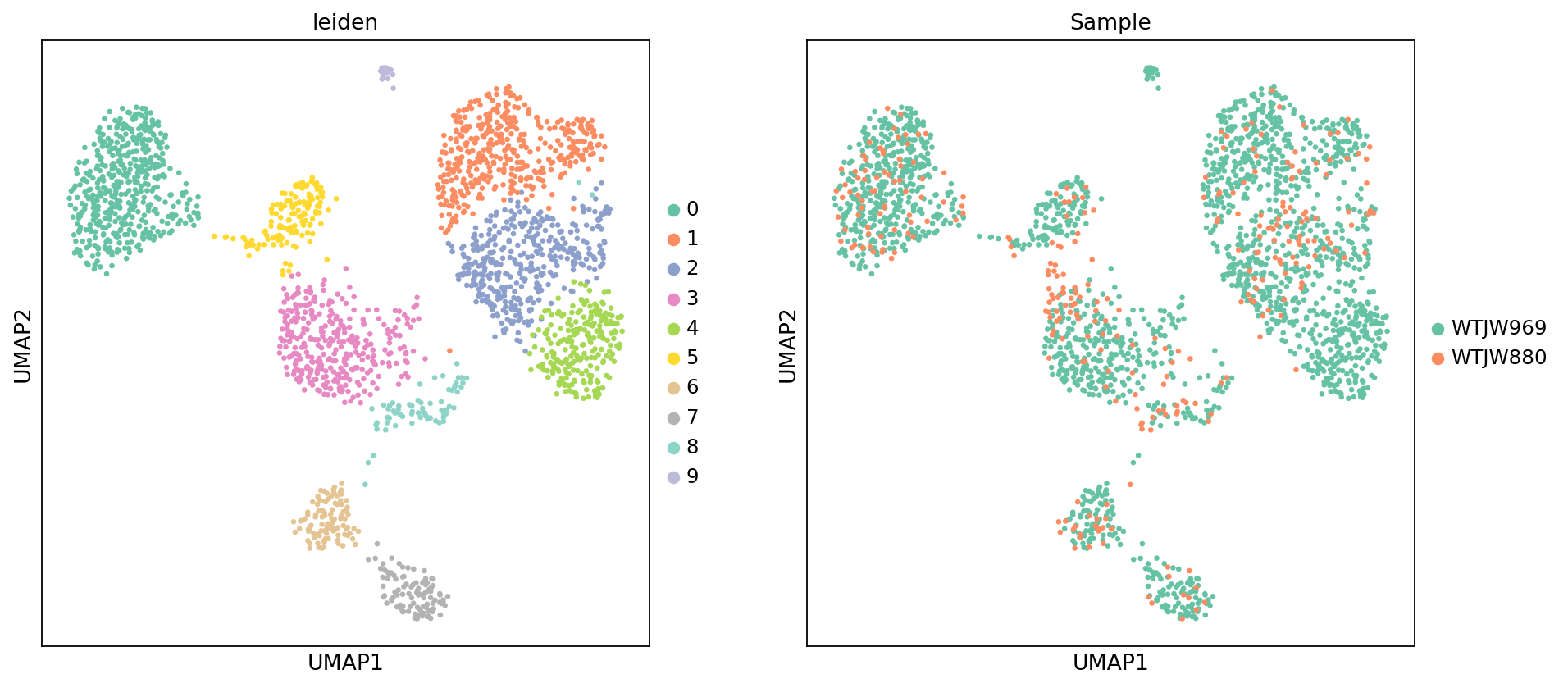
图注(甲基化 UMAP - Leiden 聚类和样本):
下图展示了甲基化数据在 UMAP 降维空间中的细胞分布,按 Leiden 聚类结果和样本 (Sample) 着色。
- 横坐标和纵坐标:UMAP 降维后的两个主维度(UMAP1 和 UMAP2),基于 LSI 降维结果计算。
- 颜色 (leiden):不同颜色代表不同的 Leiden 聚类结果,展示基于甲基化模式的细胞聚类。
- 颜色 (Sample):不同颜色代表不同的样本,用于评估样本间的批次效应。
- 点:每个点代表一个细胞。
- 用途:评估甲基化数据的聚类效果,识别基于甲基化模式的细胞亚群,以及检查样本间的批次效应。
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(6, 6))
sc.pl.umap(
adata_met,
color=['leiden', 'Sample'],
ncols=2,
s=35,
palette=my_palette
)
图注(甲基化 UMAP - 细胞类型):
下图展示了甲基化数据在 UMAP 降维空间中的细胞分布,按从 RNA 数据转移的细胞类型 (celltype) 着色。
- 横坐标和纵坐标:UMAP 降维后的两个主维度(UMAP_1 和 UMAP_2)。
- 颜色:不同颜色代表不同的细胞类型,这些细胞类型注释是从 RNA 数据转移而来的。
- 点:每个点代表一个细胞。
- 用途:评估细胞类型注释转移的效果,检查不同细胞类型在甲基化空间中的分布模式,以及验证注释的准确性。
warnings.filterwarnings('ignore')
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(6, 6))
sc.pl.umap(adata_met, color = ['celltype'], s=35, palette=my_palette)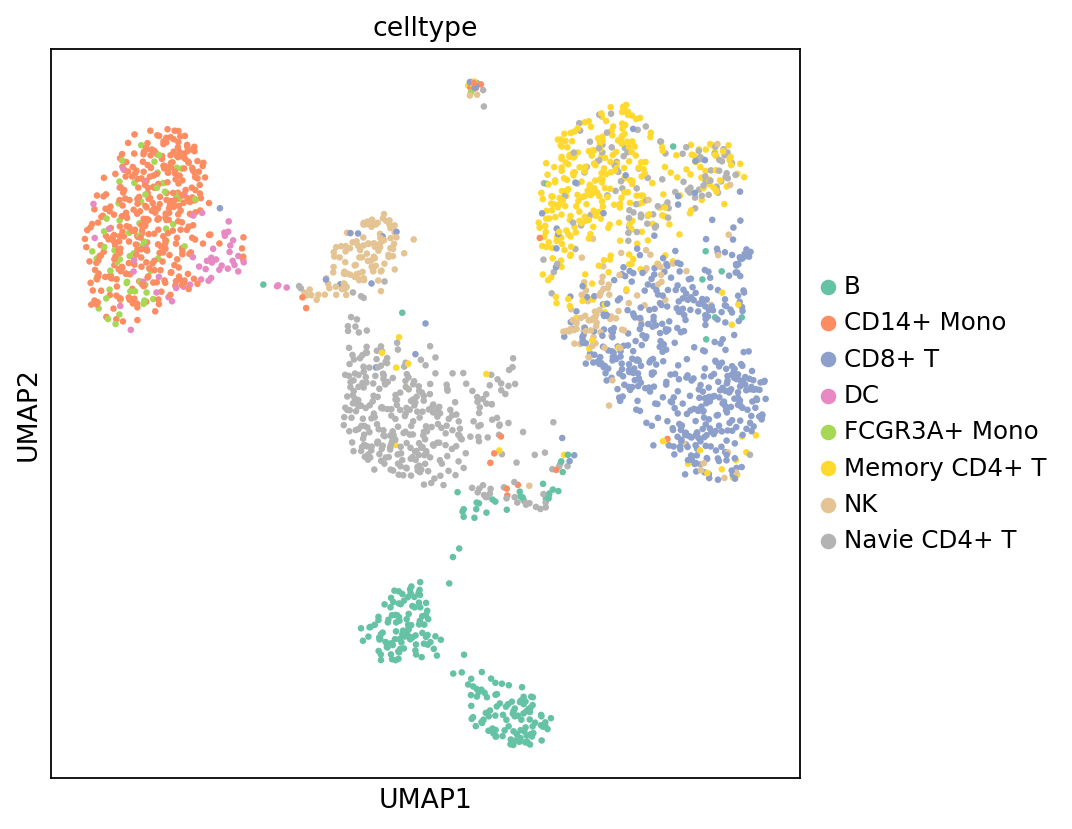
细胞组成比例
图注(细胞类型组成堆叠柱状图):
下图展示了不同样本 (Sample) 中各种细胞类型 (celltype) 的比例分布。
- 横坐标:不同样本 (Sample),便于比较不同样本的细胞类型组成。
- 纵坐标:细胞类型比例 (Proportion),范围从 0 到 1,表示各细胞类型在样本中的占比。
- 堆叠柱状图:每个柱子代表一个样本,不同颜色代表不同的细胞类型,柱子的高度表示该样本中所有细胞类型的总比例(100%)。
- 颜色:不同颜色代表不同的细胞类型,图例显示各细胞类型的名称。
- 用途:用于比较不同样本间的细胞类型组成差异,评估样本间的异质性,以及识别样本特异性的细胞类型。
def plot_bar_fraction_data_optimized(adata, x_key, y_key, custom_palette=None):
# 1. 数据准备
df = adata.obs[[x_key, y_key]].copy()
df[y_key] = df[y_key].astype('category')
df[x_key] = df[x_key].astype('category')
counts = df.groupby([x_key, y_key]).size().reset_index(name='count')
totals = counts.groupby(x_key)['count'].transform('sum')
counts['prop'] = counts['count'] / totals
prop_pivot = counts.pivot(index=x_key, columns=y_key, values='prop').fillna(0)
clusters = prop_pivot.columns.tolist()
bar_colors = None
if f'{y_key}_colors' in adata.uns:
categories = adata.obs[y_key].cat.categories
uns_colors = adata.uns[f'{y_key}_colors']
# 防止颜色数量对不上
if len(categories) == len(uns_colors):
color_dict = dict(zip(categories, uns_colors))
bar_colors = [color_dict.get(c, '#333333') for c in clusters]
# 如果上面没找到颜色,或者逻辑失败,使用自定义色盘
if bar_colors is None:
if custom_palette is None:
custom_palette = sc.pl.palettes.default_20
# 循环分配颜色
bar_colors = [custom_palette[i % len(custom_palette)] for i in range(len(clusters))]
return prop_pivot, bar_colors
# --- 绘图 ---
# 调用函数,传入你定义的 my_palette
prop_pivot, bar_colors = plot_bar_fraction_data_optimized(
adata_met,
x_key="Sample",
y_key="celltype",
custom_palette=my_palette
)
# 设置画布大小
plt.rcParams['figure.dpi'] = 100
fig, ax = plt.subplots(figsize=(5, 5))
ax.grid(False)
ax.tick_params(axis='both', which='major', labelsize=9)
# 画图
prop_pivot.plot(
kind='bar',
stacked=True,
color=bar_colors,
ax=ax,
width=0.5, # 柱子宽度
edgecolor='none',
grid=False,
zorder=3
)
# 美化坐标轴
ax.set_ylabel('Proportion', fontsize=10)
ax.set_xlabel("")
ax.tick_params(axis='x', rotation=45) # 旋转 x 轴标签
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
# 美化图例
ax.legend(
title='Cell Type',
bbox_to_anchor=(1.05, 1),
loc='upper left',
frameon=False,
fontsize=9,
title_fontsize=9,
alignment="left"
)
plt.tight_layout()
plt.show()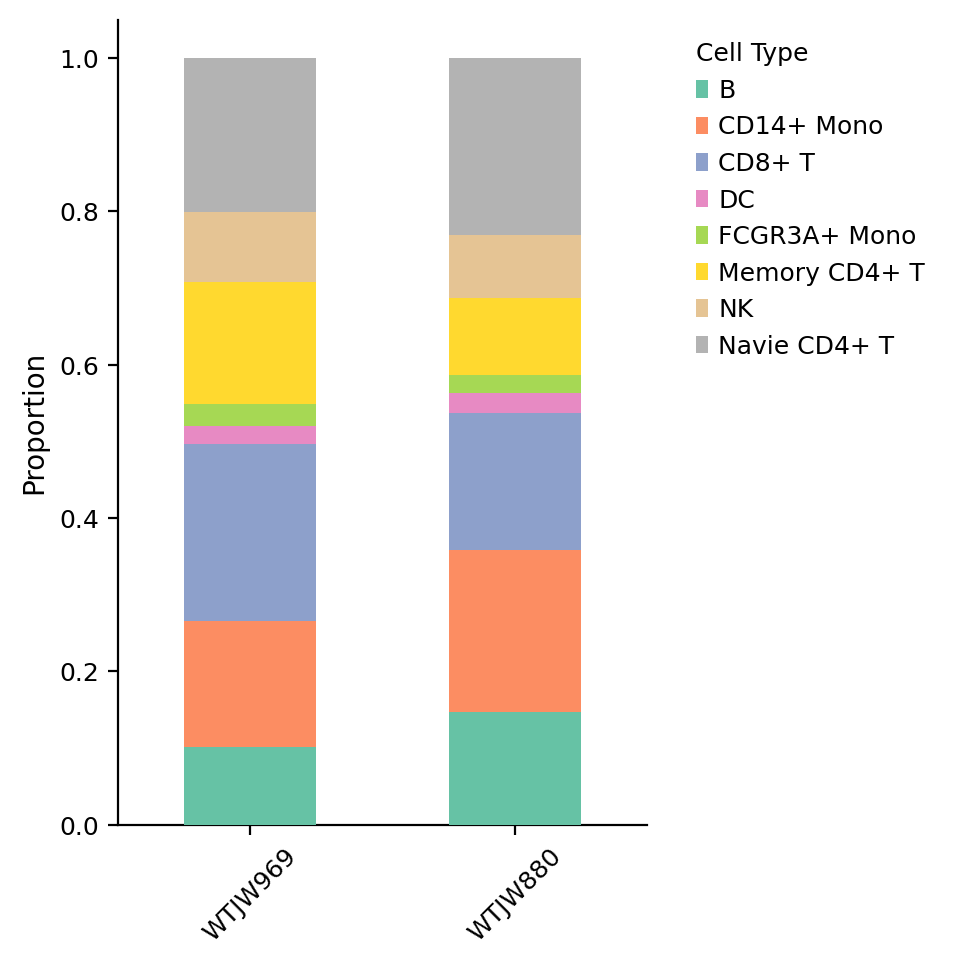
质控指标可视化
本节通过在 UMAP 空间中可视化质控指标的分布,用于评估数据整体质量及潜在异常。
图注(质控指标 UMAP 分布):
下图展示了质控指标在 UMAP 降维空间中的分布情况,用于评估数据质量。
- 横坐标和纵坐标:UMAP 降维后的两个主维度(UMAP_1 和 UMAP_2)。
- 颜色:颜色深浅表示各质控指标的数值大小,颜色越亮(黄色/白色)表示数值越高,颜色越暗(黑色/紫色)表示数值越低。
- 可视化指标:
- total_cpg_number(CpG 位点总数):反映每个细胞捕获的甲基化位点总量,数值越高表示测序深度越好。
- reads_counts(测序深度):反映每个细胞的测序深度,数值越高表示测序数据越多。
- genome_cov(基因组覆盖度):量化测序数据在基因组区域的覆盖比例,数值越高表示覆盖越全面。
- CpG%(CpG 甲基化率):展现整体 CpG 位点的甲基化水平,通常在 0-100% 范围内。
- CH%(CH 非 CpG 甲基化率):揭示非经典甲基化位点的修饰水平,哺乳动物细胞通常 < 5%。
- 点:每个点代表一个细胞。
- 用途:用于识别质控指标的空间分布模式,发现异常细胞(如质控指标异常高或低的细胞),以及评估数据质量的均匀性。
# --- 质控指标 UMAP 可视化 ---
plt.rcParams['font.size'] = 10
plt.rcParams['axes.titlesize'] = 10
# 可视化 total_cpg_number、reads_counts、genome_cov、CpG%、CH%
features = ['total_cpg_number', 'reads_counts', 'genome_cov','CpG%', 'CH%']
sc.pl.umap(
adata_met,
color=features,
ncols=3,
color_map=my_warm_cmap,
vmax='p99',
s=8,
frameon=False,
vmin=0,
show=False
)
plt.gcf().set_size_inches(10, 5)
plt.show()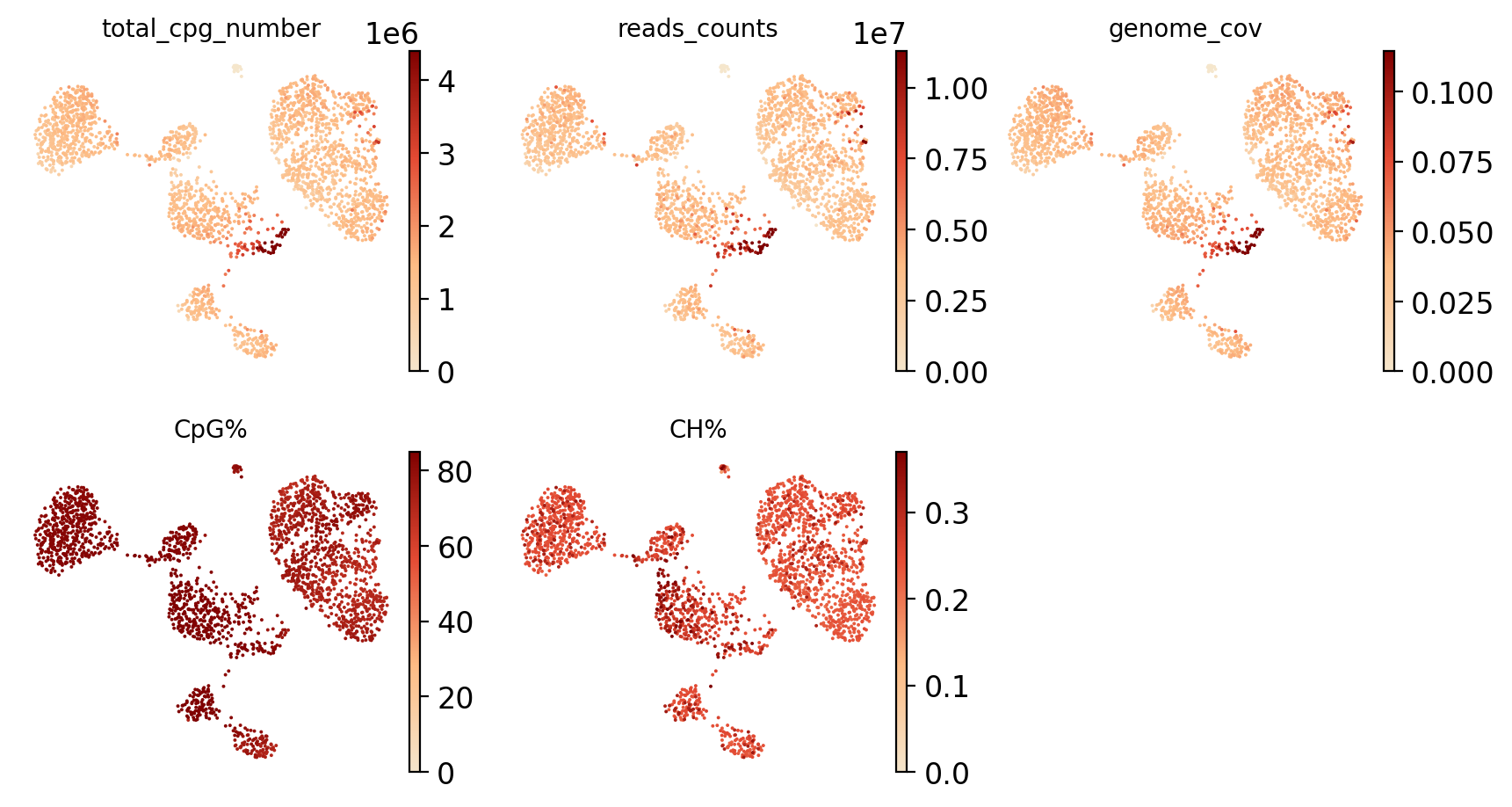
质控指标小提琴图可视化
本节使用小提琴图展示质控指标在不同细胞分组(Leiden 聚类和细胞类型)中的分布特征,用于评估数据质量在不同细胞群体中的一致性。
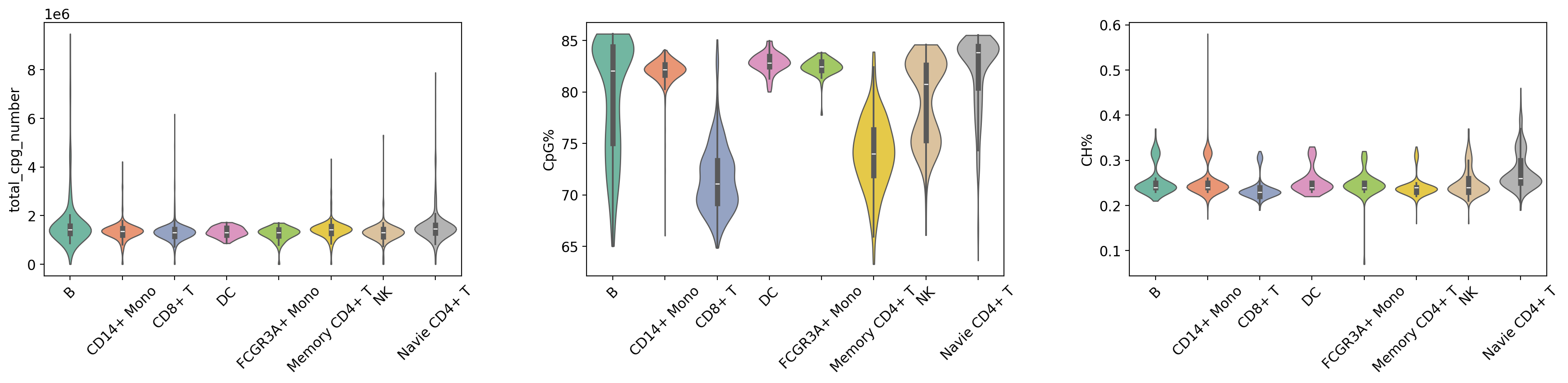
图注(质控指标小提琴图):
下图展示了质控指标在不同细胞分组(Leiden 聚类和细胞类型)中的分布特征。
- 横坐标:不同的细胞分组,包括 Leiden 聚类 (leiden) 和细胞类型 (celltype),便于比较不同细胞分组的质控指标差异。
- 纵坐标:各质控指标的数值,包括
total_cpg_number(CpG 位点总数)、CpG%(CpG 甲基化率)和CH%(CH 甲基化率)。- 小提琴图:每个小提琴图代表一个细胞分组,宽度表示该数值区间的细胞密度,越宽表示该区间的细胞越多。小提琴图还包含箱线图,显示中位数、四分位数和异常值。
- 分组方式:
- 按 Leiden 聚类分组:展示不同聚类间的质控指标差异,用于评估聚类质量。
- 按细胞类型分组:展示不同细胞类型间的质控指标差异,用于评估细胞类型注释的合理性。
- 用途:用于评估数据质量在不同细胞分组中的均匀性,识别质控指标异常的细胞组,以及验证聚类和注释结果的可靠性。
warnings.filterwarnings('ignore')
plt.rcParams['figure.figsize'] = (6, 4)
plt.rcParams['axes.grid'] = False
violin_params = {
'palette': my_palette,
'stripplot': False,
'inner': 'box',
'linewidth': 1,
'size': 1,
'cut': 0,
'multi_panel': True
}
print("\n=== 质控指标分布 (按 Cluster) ===")
# 按 leiden 聚类分组
sc.pl.violin(
adata_met,
keys=['total_cpg_number', 'CpG%', 'CH%'],
groupby='leiden',
**violin_params
)
plt.show()
print("\n=== 质控指标分布 (按 Cell Type) ===")
# 按细胞类型分组 (加了旋转)
sc.pl.violin(
adata_met,
keys=['total_cpg_number', 'CpG%', 'CH%'],
groupby='celltype',
rotation=45,
**violin_params
)
plt.show()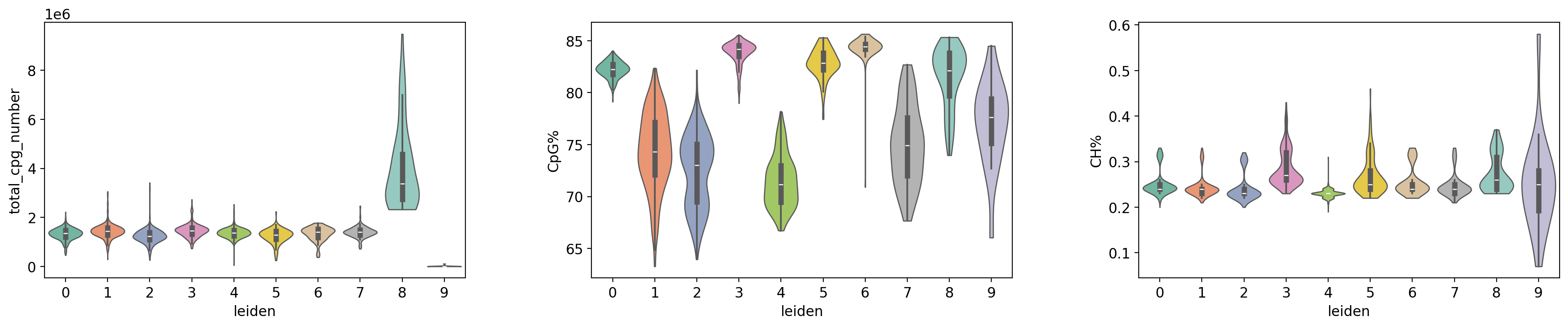
=== 质控指标分布 (按 Cell Type) ===
adata_met.write_h5ad('adata_met.h5ad')