甲基化 + RNA 双组学: CNV 拷贝数变异分析 (基于 CopyKit)
模块简介
本模块基于 CopyKit (v0.1.3) 开发,专为从单细胞测序数据中推断全基因组拷贝数变异 (CNV) 而设计。
CopyKit 是一套功能强大的 R 语言工具集,能够对单细胞数据进行系统性的预处理、标准化、分段 (Segmentation) 和聚类分析。通过检测基因组范围内的覆盖度 (Coverage) 变化,实现以下核心分析目标:
- 变异识别 (Variation Detection):精准定位肿瘤细胞中的染色体扩增 (Amplification) 与缺失 (Deletion) 事件,绘制全基因组 CNV 图谱。
- 细胞分群 (Cell Classification):基于 CNV 特征,有效区分正常细胞(二倍体)与肿瘤细胞(非二倍体/非整倍体)。
- 克隆进化 (Clonal Evolution):通过亚克隆结构重构,探索肿瘤内部的异质性 (Intra-tumor Heterogeneity) 及克隆进化轨迹。
注意: 本分析主要适用于人源肿瘤样本,依赖于人类参考基因组(hg19/hg38)进行比对和注释。
输入文件准备
本模块支持两种类型的输入文件,请根据您的数据处理阶段选择:
方案 A:直接导入已分析结果 (推荐)
如果您之前已经运行过 CopyKit 的 runVarbin 流程,可以直接使用生成的 .rds 文件。这是最快捷的方式。
文件结构示例:
workspace/data/AY1768876207519/methylation/demo3MDA-task-2/MDA_cs/cnvPreflight/
├── copykit_MDA_cs_1Mb.rds # 1Mb 分辨率结果
├── copykit_MDA_cs_500kb.rds # 500kb 分辨率结果
└── copykit_MDA_cs_220kb.rds # 220kb 分辨率结果方案 B:从单细胞 BAM 文件开始
如果您拥有拆分后的单细胞 BAM 文件,可以直接作为输入。
注意:输入的 BAM 文件必须经过 Flag 矫正 和 去重 (Deduplication) 处理,以确保定量准确性。
文件结构示例:
./
├── AAAGAAGAAGAGTTTAG.dedup.bam
├── AAAGAAGAAGGAGTGGT.dedup.bam
├── ...
└── TTTGGATATTTGTGAGG.dedup.bam详细教程:关于如何生成上述标准的单细胞 BAM 文件(包括拆分、Flag 矫正、去重),请参考完整指南: 指南
CopyKit 分析流程
关键参数设置
在分析过程中,以下三个参数对于数据质量控制和结果分辨率至关重要:
| 参数名称 | 中文释义 | 默认值 | 详细说明 |
|---|---|---|---|
bin_sizes | 基因组窗口大小 | 220kb, 500kb, 1Mb | 决定 CNV 分析的分辨率。 • 越小 (e.g. 220kb):分辨率越高,能发现更微小的变异,但对测序深度要求更高,噪音可能增加。 • 越大 (e.g. 1Mb):数据更平滑,噪音更低,但只能发现大片段变异。 注意:修改此参数需要重新运行 runVarbin 步骤,该步骤输入为 BAM 文件且耗时较长。 |
min_bincount | 最小 Reads 丰度 | 10 | 质量控制 (QC) 参数。 要求每个细胞在每个 Bin 中的平均 Reads 数必须大于此值。用于剔除测序深度不足、无法准确推断 CNV 的低质量细胞。 |
pearson_corr_cutoff | pearson 相关性过滤阈值 | 0.8 (软件默认 0.9) | 质量控制 (QC) 参数(请勿与 bin_size 混淆)。CopyKit 会计算每个细胞与其最近邻(k=5)细胞的 Pearson 相关性。如果相关性低于此阈值,该细胞被视为“离群值”或低质量细胞并被过滤。 • 提高此值会获得更纯净的克隆,但可能丢失部分异质性细胞。 |
最佳实践建议
关于
bin_sizes的选择:- 本模块默认同时计算 220kb, 500kb, 和 1Mb 三种分辨率。
- CopyKit 支持的分辨率列表:55kb, 110kb, 195kb, 220kb, 280kb, 500kb, 1Mb, 2.8Mb。
- 策略:如果样本测序深度较低,建议优先参考 1Mb 或 500kb 的结果;深度较高时可使用 220kb 以获得更精细的结果。
- CopyKit 内部已预设并优化了这些 Bin 区域,过滤了重复序列及比对困难区域。
关于过滤参数 (
min_bincount&resolution):- 如果保留的细胞过少,可适当降低
min_bincount(例如降至 5),但这会增加噪声。 - 如果聚类结果中包含大量杂乱的单细胞,可适当提高
pearson_corr_cutoff阈值(例如升至 0.9)。
- 如果保留的细胞过少,可适当降低
library(copykit)
library(BiocParallel)
library(ggplot2)
# --- 输入参数配置 ---
## genome_version:数据比对的参考基因组版本
genome_version <- 'hg38'
## outdir:结果输出目录
outdir <- '/PROJ2/FLOAT/weiqiuxia/project/20241227_methy/script/standard_reports/modules/copykit/test/results/MDA_cs'
## bam_dir:单细胞 BAM 文件所在目录 (方案 B)
bam_dir <- NULL
## rds_path:已生成的 RDS 文件目录 (方案 A)
rds_path <- '/PROJ2/FLOAT/weiqiuxia/project/20241227_methy/script/standard_reports/modules/copykit/test/input/MDA_cs/'
## samplename:样本名称标识
samplename <- 'MDA_cs'
## min_bincount:最小平均 Reads 数过滤阈值
# 用于过滤掉 Read Counts 较低的低质量细胞,建议保持默认值
min_bincount <- 10
## bin_sizes:分析分辨率列表
bin_sizes <- c('220kb', '500kb', '1Mb')
## pearson_corr_cutoff:相关性过滤阈值 (用于 findOutliers)
pearson_corr_cutoff <- 0.8
## is_paired_end:是否为双端测序 (TRUE/FALSE)
is_paired_end <- TRUE
## remove_Y:是否移除 Y 染色体 (TRUE/FALSE)
remove_Y <- TRUE
## threads:并行计算线程数
threads <- 8
## skipKNNSmooth:是否跳过KNNSmooth(TRUE/FALSE)
skipKNNSmooth <- FALSE
dir.create(outdir, recursive = TRUE)plotConsensusLine_static <- function(scCNA, sample) {
# bindings for NSE
start <- xstart <- xend <- abspos <- NULL
####################
## checks
####################
if (nrow(consensus(scCNA)) == 0) {
stop("Slot consensus is empty. Run calcConsensus()")
}
####################
## aesthetic setup
####################
# obtaining chromosome lengths
chr_ranges <-
as.data.frame(SummarizedExperiment::rowRanges(scCNA))
chr_lengths <- rle(as.numeric(chr_ranges$seqnames))$lengths
# obtaining first and last row of each chr
chr_ranges_start <- chr_ranges %>%
dplyr::group_by(seqnames) %>%
dplyr::arrange(seqnames, start) %>%
dplyr::filter(dplyr::row_number() == 1) %>%
dplyr::ungroup()
chr_ranges_end <- chr_ranges %>%
dplyr::group_by(seqnames) %>%
dplyr::arrange(seqnames, start) %>%
dplyr::filter(dplyr::row_number() == dplyr::n()) %>%
dplyr::ungroup()
# Creating data frame object for chromosome rectangles shadows
chrom_rects <- data.frame(
chr = chr_ranges_start$seqnames,
xstart = as.numeric(chr_ranges_start$abspos),
xend = as.numeric(chr_ranges_end$abspos)
)
xbreaks <- rowMeans(chrom_rects %>%
dplyr::select(
xstart,
xend
))
if (nrow(chrom_rects) == 24) {
chrom_rects$colors <- rep(
c("white", "gray"),
length(chr_lengths) / 2
)
} else {
chrom_rects$colors <- c(rep(
c("white", "gray"),
(length(chr_lengths) / 2)
), "white")
}
# Creating the geom_rect object
ggchr_back <-
list(geom_rect(
data = chrom_rects,
aes(
xmin = xstart,
xmax = xend,
ymin = -Inf,
ymax = Inf,
fill = colors
),
alpha = .2
), scale_fill_identity())
sec_breaks <- c(0, 0.5e9, 1e9, 1.5e9, 2e9, 2.5e9, 3e9)
sec_labels <- c(0, 0.5, 1, 1.5, 2, 2.5, 3)
# theme
ggaes <- list(
scale_x_continuous(
breaks = xbreaks,
labels = gsub("chr", "", chrom_rects$chr),
position = "top",
expand = c(0, 0),
sec.axis = sec_axis(
~.,
breaks = sec_breaks,
labels = sec_labels,
name = "genome position (Gb)"
)
),
theme_classic(),
theme(
axis.text.x = element_text(
angle = 0,
vjust = .5,
size = 15
),
axis.text.y = element_text(size = 15),
panel.border = element_rect(
colour = "black",
fill = NA,
size = 1.3
),
legend.position = "top",
axis.ticks.x = element_blank(),
axis.title = element_text(size = 15),
plot.title = element_text(size = 15)
)
)
####################
# obtaining and wrangling data
####################
con <- consensus(scCNA)
con_l <- con %>%
dplyr::mutate(abspos = chr_ranges$abspos) %>%
tidyr::gather(
key = "group",
value = "segment_ratio",
-abspos
)
choice <- gtools::mixedsort(unique(con_l$group))
df_plot <- con_l %>% dplyr::filter(group %in% sample)
p <- ggplot(df_plot) +
ggchr_back +
ggaes +
geom_line(aes(abspos, segment_ratio, color = group),size = 1.2, alpha = .7) +
labs(
x = "",
y = "consensus segment ratio"
)
return(p)
}# 设置并行参数
register(MulticoreParam(progressbar = T, workers = threads), default = T)
# --- 输入路径逻辑判断 ---
# 优先读取 rds_path 下的数据,若不存在则尝试读取 bam_dir
bam_flag <- FALSE
if(!is.null(bam_dir) & !is.null(rds_path)){
if(!dir.exists(rds_path)){
if(!dir.exists(bam_dir)){
stop("Error: Both bam_dir and rds_path do not exist!")
}else{
input_dir <- bam_dir
bam_flag <- TRUE
}
}else{
input_dir <- rds_path
}
} else if(is.null(bam_dir) & !is.null(rds_path)){
input_dir <- rds_path
} else if(!is.null(bam_dir) & is.null(rds_path)){
input_dir <- bam_dir
bam_flag <- TRUE
} else{
stop("Error: Please set either bam_dir or rds_path!")
}
# --- 基因组版本识别 ---
if(length(grep('(GRCh38)|(hg38)', genome_version, ignore.case = TRUE )) > 0){
genome <- 'hg38'
}else if(length(grep('(GRCh37)|(hg19)', genome_version, ignore.case = TRUE )) > 0){
genome <- 'hg19'
}else{
stop('Error: Cannot recognize genome version. Please use hg38, GRCh38, hg19, or GRCh37.')
}
# --- 运行 runVarbin (如果输入是 BAM) ---
if(bam_flag){
for(i in bin_sizes){
message(paste("Processing bin size:", i))
tumor <- runVarbin(input_dir,
min_bincount = min_bincount,
remove_Y = remove_Y,
is_paired_end = is_paired_end,
resolution = i,
genome = genome)
saveRDS(tumor, paste0(outdir, '/copykit_', samplename, '_', i, '.rds'))
}
input_dir <- outdir
}数据预处理原理
CopyKit 使用核心函数 runVarbin 对输入的 BAM 文件进行预处理。该过程包含以下关键模块:
runCountReads(分箱计数):- Bin Counting:基于预设的基因组窗口 (Bins),使用 featureCounts 统计落在每个 Bin 内的 Reads 数。
- 去重:统计时会自动忽略被标记为 PCR 重复 (PCR Duplicates) 的 Reads。
- 双端处理:若
is_paired_end = TRUE,则将双端 Reads 作为一个 Fragment 进行计数。 - 过滤:默认移除 Y 染色体区域(可选),并根据计数结果剔除平均 Read Counts 过低的低质量细胞。
- GC 校正:理论上测序深度应均匀分布,但实际上高 GC 含量区域往往存在 PCR 扩增偏差,导致 Fragment 计数与 GC 含量呈正相关。在 CNV 分析前,算法会对每个细胞进行 GC 偏差校正,将受 GC 影响的计数拉回基准线,以消除系统误差。
runVst(方差稳定变换):- 原理:单细胞 DNA 测序中的 Bin Counts 通常服从泊松分布 (Poisson Distribution),其特点是方差等于均值。这导致高丰度区域的噪声远大于低丰度区域。
- 作用:使用 Freeman-Tukey (FT) 变换将数据转换为近似正态分布。变换后的数据方差近似恒等于 1,且不再依赖于均值。这一步对于后续准确检测断点 (Segmentation) 至关重要。
runSegmentation(CBS 分割):- 平滑:首先使用平滑算法修剪掉极少数远高于或低于邻近 Bin 的“尖刺”数值,减少噪声干扰。
- CBS 算法:对每个细胞运行 Circular Binary Segmentation (CBS) 算法,在基因组上寻找拷贝数发生变化的断点 (Breakpoints)。
- 合并:将基因组上连续且计数相似的 Bins 合并为 Segments。同一 Segment 内的所有 Bins 具有相同的 Segment Ratio。
- Segment Ratio 含义 (相对于平均倍性):
1.0:二倍体状态 (Diploid)。1.5:3 拷贝 (Gain)。0.5:单拷贝丢失 (Loss)。2.0:4 拷贝扩增 (Amplification)。
logNorm(对数归一化):- 对 Segment Ratio 进行 Log 转换,生成
logr矩阵,便于后续 UMAP 降维聚类分析。
- 对 Segment Ratio 进行 Log 转换,生成
运行完runVarbin生成的矩阵 (Bin x Cells):
bincounts:GC 校正后的原始 Read Counts 矩阵。ft:经过方差稳定变换 (VST) 后的数值矩阵,作为分割算法的输入。smoothed_bincounts:剔除离群点后的平滑计数,用于绝对拷贝数推断。segment_ratios:分割后的比率矩阵,是细胞过滤、绘图、分群及 CNV 分析的核心数据。ratios:基于 smoothed_bincounts 归一化后的比率矩阵。logr:Log 转换后的 Segment Ratio。
以上矩阵可通过
assay(tumor_list[['220kb']], 'bincounts')形式获取
varbin_rds <- list.files(rds_path,pattern = '.rds', full.names = T)质量控制
runMetrics 函数基于 bincounts 计算每个细胞的 Overdispersion (过度离散度),同时我们会可视化每个细胞的 Reads 总数、 能分配到 Bin 的 Reads 总数以及 Breakpoints 数目。
关键质控指标
- Overdispersion (过度离散度):
- 反映测序覆盖的均一度。理论上相邻 Bin 的 Read Counts 应相近。
- 若 Overdispersion 值过高,说明相邻 Bin 差异大,技术噪声明显,可能影响 CNV 结果的准确性。
- Breakpoint Count (断点数):
- 正常的二倍体细胞,或未发生变异的细胞,其 Breakpoint Count 通常接近于 0。
- 肿瘤细胞由于基因组不稳定,通常具有较高的断点数。
- Total Reads(总 Reads 数):
- 每个细胞的 Reads 数目,可以反应每个细胞的数据量是否充足。
- Reads Assigned bins(能分配到 Bin 的 Reads 数目):
- 有效 Reads 数。如果此数值过低,说明大量 Reads 比对到了重复序列或低质量区域(CopyKit 预设的 Bins 已经过滤了这些区域),提示细胞测序质量不佳。
CopyKit 建议:通常 1M reads/cell 的测序深度和约 10% 的 Duplication 比例,即可获得高质量的 220kb 分辨率 CNV 结果。
以下展示了不同分辨率下的质控指标分布:
图注:下图展示了四个质控指标的分布。
- 横坐标:样本。
- 纵坐标:各指标的具体数值。
- 颜色:代表
overdispersion(过度离散度)的大小,颜色越亮代表数据噪声越大。- 判读:高质量数据通常表现为较高的
reads_assigned_bins和较低的overdispersion。
tumor_list <- list()
for(i in varbin_rds){
bin_size_name <- gsub('.*_(\\d+[Mk]b).rds','\\1',i)
tumor_list[[bin_size_name]] <- readRDS(i)
tumor_list[[bin_size_name]] <- runMetrics(tumor_list[[bin_size_name]])
}|======================================================================| 100%
Counting breakpoints.
Done.
Calculating overdispersion.
|======================================================================| 100%
Counting breakpoints.
Done.
Calculating overdispersion.
|======================================================================| 100%
Counting breakpoints.
Done.
options(repr.plot.width = 8, repr.plot.height = 7, repr.plot.res = 150)
pic <- plotMetrics(
tumor_list[['220kb']],
metric = c(
"reads_total",
"reads_assigned_bins",
"overdispersion",
"breakpoint_count"),
label = 'overdispersion') +
ggtitle('220kb Metrics') +
theme(plot.title = element_text(hjust = .5, size = 20))
pic
ggsave(filename = file.path(outdir, paste0('plotMetrics_',samplename, '_220kb.pdf')), pic, width = 8, height = 7)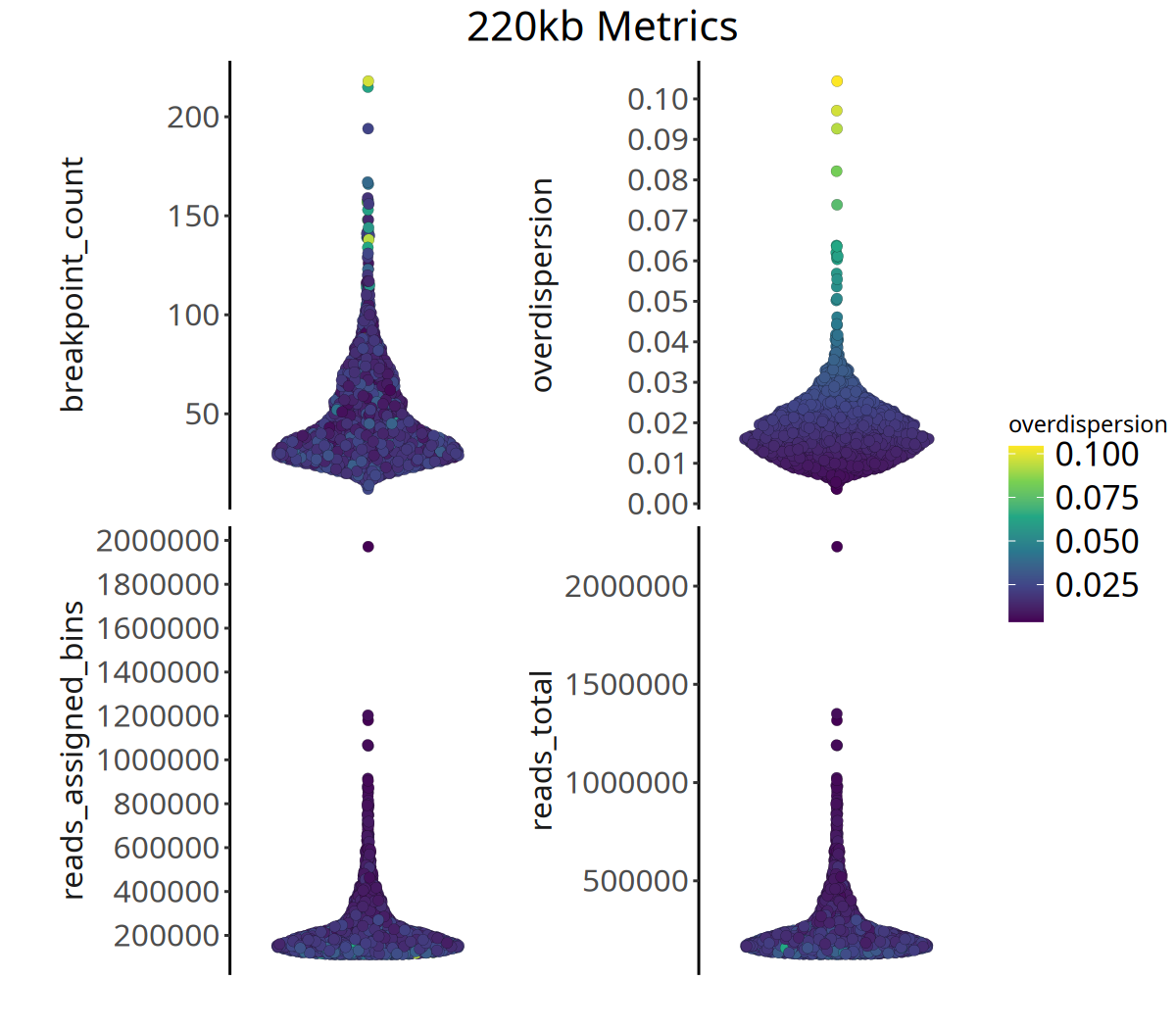
options(repr.plot.width = 8, repr.plot.height = 7, repr.plot.res = 150)
pic <- plotMetrics(
tumor_list[['500kb']],
metric = c(
"reads_total",
"reads_assigned_bins",
"overdispersion",
"breakpoint_count"),
label = 'overdispersion') +
ggtitle('500kb Metrics') +
theme(plot.title = element_text(hjust = .5, size = 20))
pic
ggsave(filename = file.path(outdir, paste0('plotMetrics_',samplename, '_500kb.pdf')), pic, width = 8, height = 7)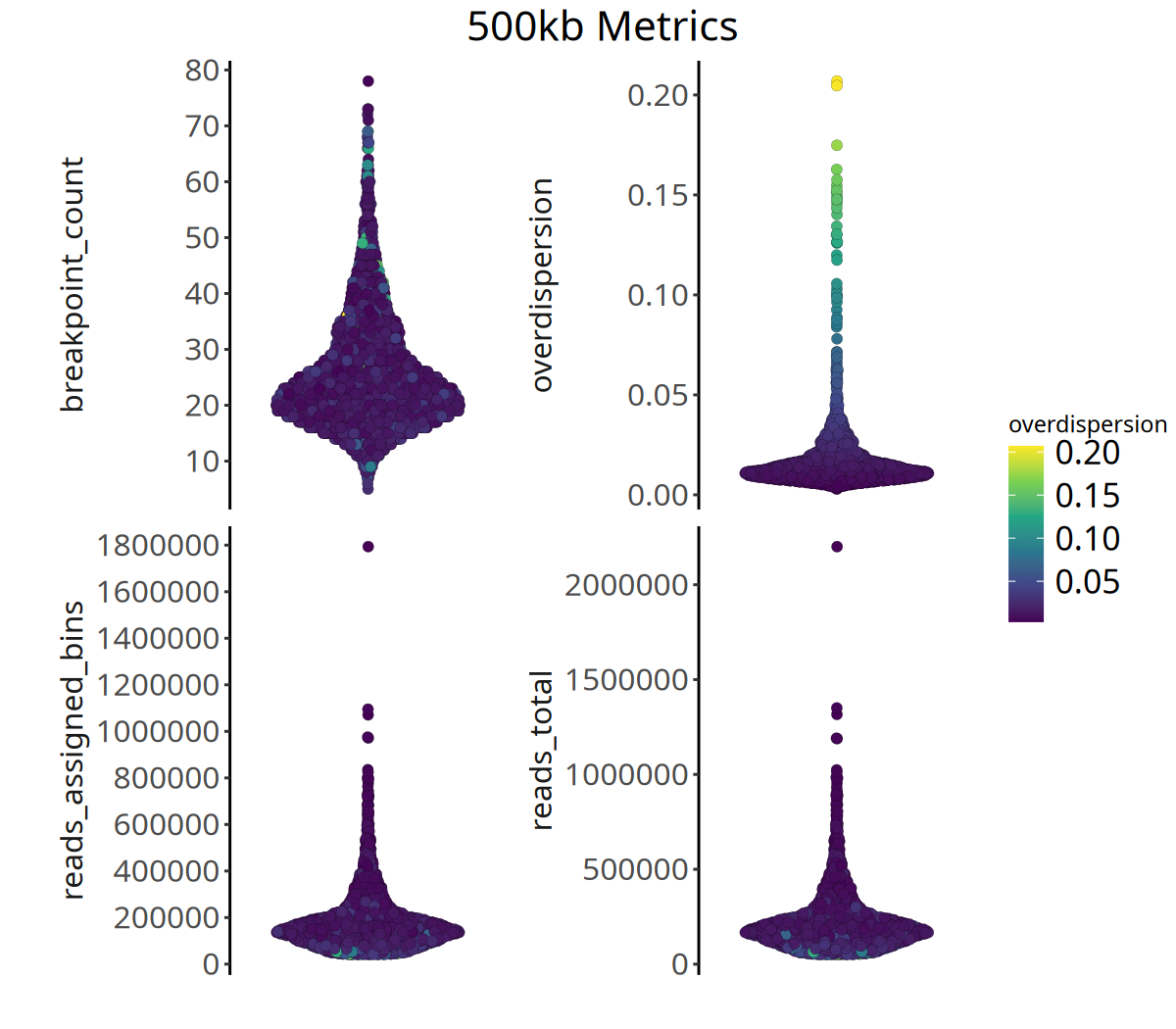
options(repr.plot.width = 8, repr.plot.height = 7, repr.plot.res = 150)
pic <- plotMetrics(
tumor_list[['1Mb']],
metric = c(
"reads_total",
"reads_assigned_bins",
"overdispersion",
"breakpoint_count"),
label = 'overdispersion') +
ggtitle('1Mb Metrics') +
theme(plot.title = ggplot2::element_text(hjust = .5, size = 20))
pic
ggsave(filename = file.path(outdir, paste0('plotMetrics_',samplename, '_1Mb.pdf')), pic, width = 8, height = 7)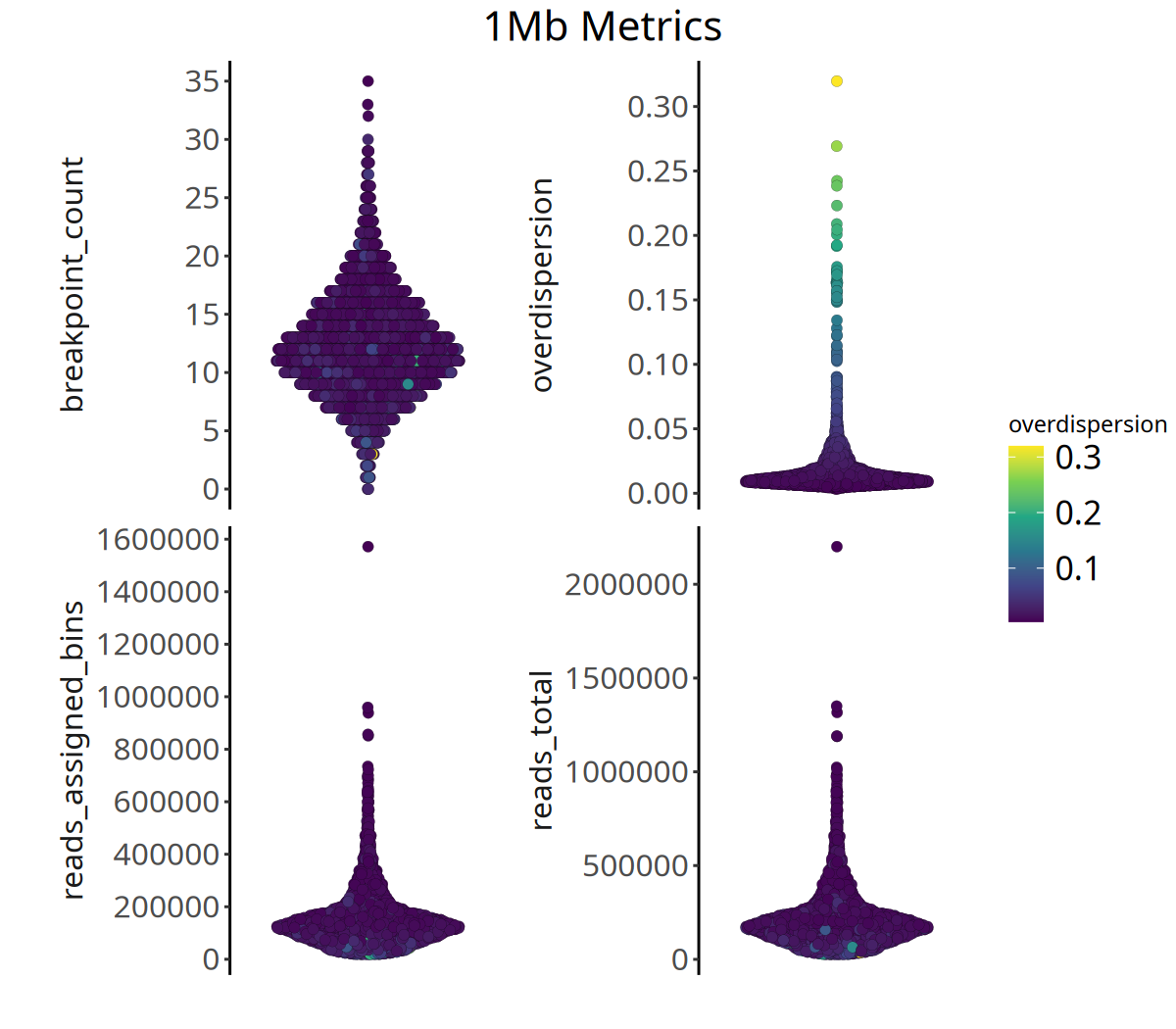
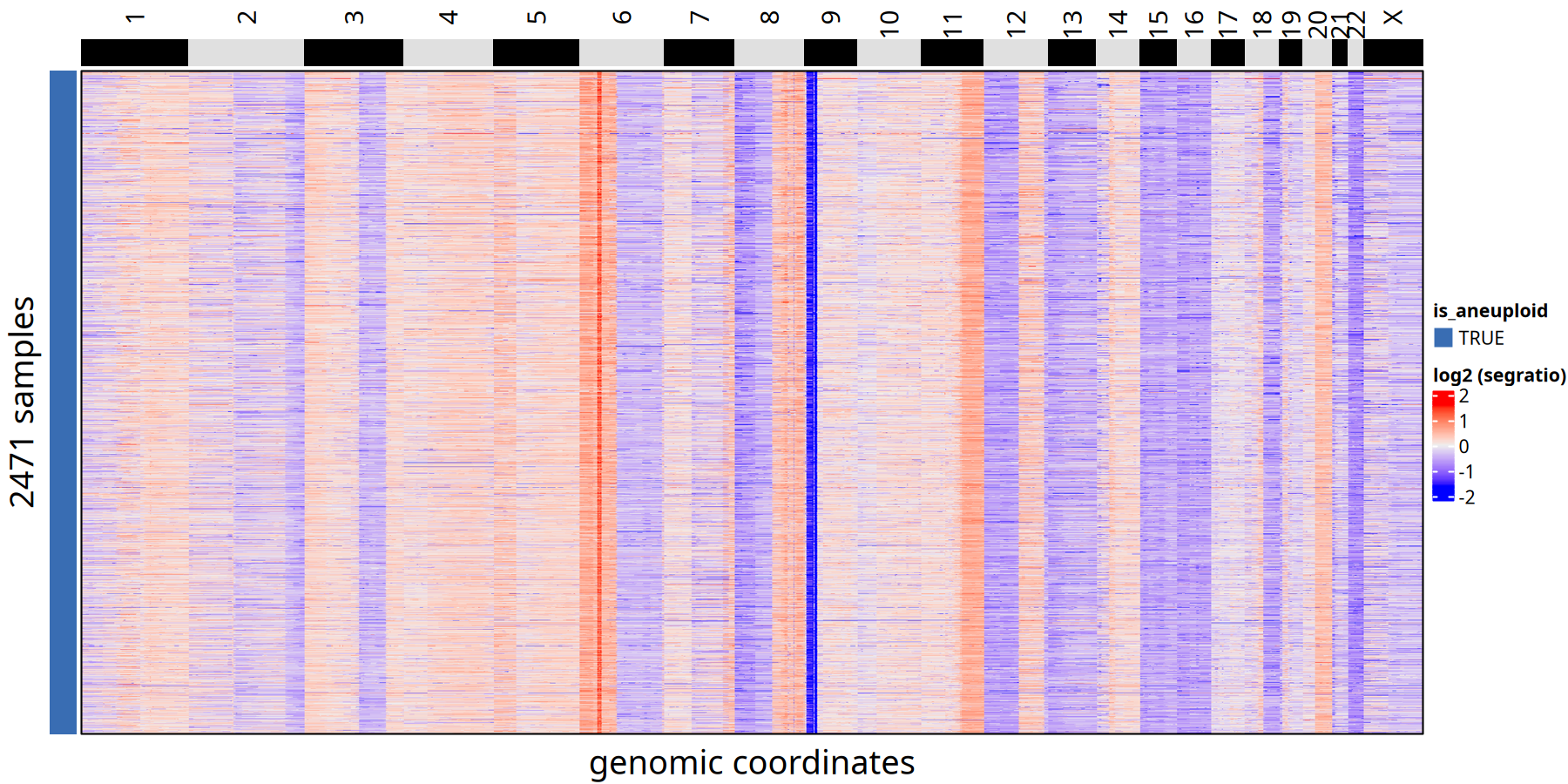
正常二倍体细胞过滤
在进行克隆演化分析、聚类和谱系树构建之前,必须剔除正常的二倍体细胞。如果保留这些细胞,它们会聚集成一个巨大的“假克隆”,严重干扰对肿瘤异质性的解析。
findAneuploidCells 函数用于区分正常细胞和非整倍体(肿瘤)细胞:
- 判断依据:正常二倍体细胞的 Segment Ratios 应为一条平稳直线,其变异系数 (CV = SD / Mean) 接近于 0。
- 肿瘤细胞:由于存在大量扩增和缺失,其 Segment Ratios 标准差较大,CV 值较高。
- 双峰分布:正常细胞和肿瘤细胞的 CV 分布通常呈现双峰模式。如果一个细胞的 CV 波动超过二倍体背景噪声的 5 倍,则被判定为非整倍体 (Aneuploid)。
220kb bin 的 diploid 和 aneuploid 的 CNV 结果
图注说明: 以下热图中,每一列代表基因组上的一个 Bin (按位置排序),每一行代表一个细胞。
- 左侧颜色条 (
is_aneuploid):TRUE表示判定为非整倍体(肿瘤)细胞,FALSE表示正常二倍体细胞。- 热图颜色:代表 Log2 转化的 Segment Ratio。红色表示拷贝数扩增 (Gain/Amp),蓝色表示拷贝数缺失 (Loss/Del),白色表示二倍体状态 (Neutral)。
options(repr.plot.width = 12, repr.plot.height = 6, repr.plot.res = 150)
suppressMessages({
tumor_list[['220kb']] <- findAneuploidCells(tumor_list[['220kb']])
ht <- plotHeatmap(
tumor_list[['220kb']],
label = 'is_aneuploid',
row_split = 'is_aneuploid',
n_threads = threads)
ht
})
pdf(file.path(outdir, paste0('plotHeatmap_', samplename, '_220k_aneuploid.pdf')), width = 12, height = 6)
ht
dev.off()pdf: 2
500kb bin 的 diploid 和 aneuploid 的 CNV 结果
options(repr.plot.width = 12, repr.plot.height = 6, repr.plot.res = 150)
suppressMessages({
tumor_list[['500kb']] <- findAneuploidCells(tumor_list[['500kb']])
ht <- plotHeatmap(tumor_list[['500kb']], label = 'is_aneuploid', row_split = 'is_aneuploid', n_threads = threads)
})
pdf(file.path(outdir, paste0('plotHeatmap_', samplename, '_500k_aneuploid.pdf')), width = 12, height = 6)
ht
dev.off()pdf: 2
1Mb bin 的 diploid 和 aneuploid 的 CNV 结果
options(repr.plot.width = 12, repr.plot.height = 6, repr.plot.res = 150)
suppressMessages({
tumor_list[['1Mb']] <- findAneuploidCells(tumor_list[['1Mb']])
ht <- plotHeatmap(tumor_list[['1Mb']], label = 'is_aneuploid', row_split = 'is_aneuploid', n_threads = threads)
})
pdf(file.path(outdir, paste0('plotHeatmap_', samplename, '_1Mb_aneuploid.pdf')), width = 12, height = 6)
ht
dev.off()pdf: 2
# 过滤掉正常二倍体细胞
for(i in names(tumor_list)){
tumor_list[[i]] <- tumor_list[[i]][,colData(tumor_list[[i]])$is_aneuploid == TRUE]
}离群细胞过滤
为了构建准确的肿瘤亚克隆和进化关系,我们需要进一步使用 findOutliers 剔除那些与其他细胞相关性极低、可能是技术噪声或质量不佳的细胞。
该步骤使用 Pearson 相关性作为衡量标准。如果一个细胞与其 K 个最近邻居的平均相关性低于 pearson_corr_cutoff (默认 0.9,本教程设置为 0.8),则被标记为 outlier。
for(i in names(tumor_list)){
tumor_list[[i]] <- findOutliers(tumor_list[[i]], resolution = pearson_corr_cutoff)
}|======================================================================| 100%
Marked 1469 cells as outliers.
Adding information to metadata. Access with colData(scCNA).
Done.
Calculating correlation matrix.
|======================================================================| 100%
Marked 1144 cells as outliers.
Adding information to metadata. Access with colData(scCNA).
Done.
Calculating correlation matrix.
|======================================================================| 100%
Marked 1493 cells as outliers.
Adding information to metadata. Access with colData(scCNA).
Done.
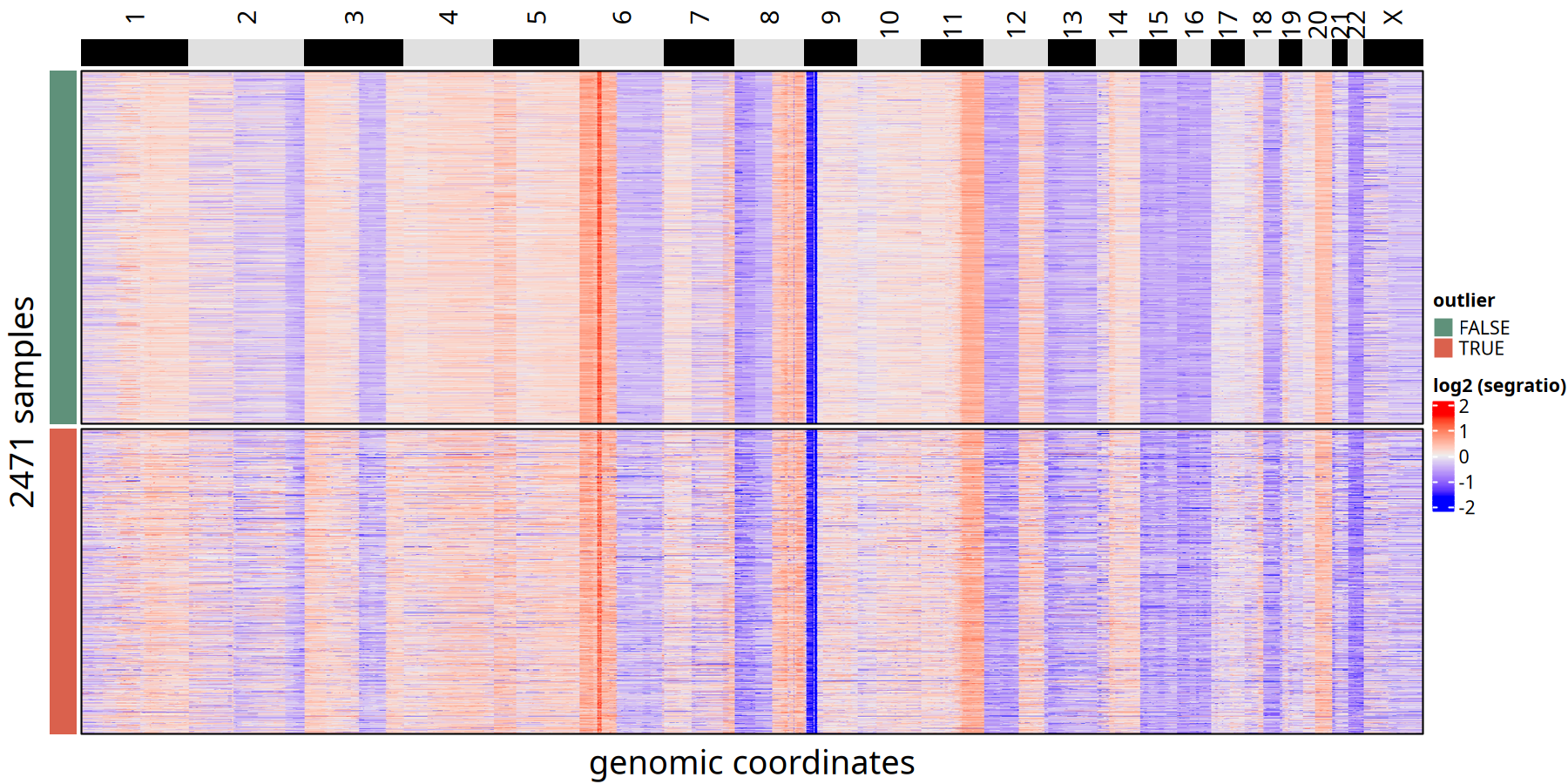
220kb 分辨率下离群细胞与保留细胞的 CNV 热图对比
图注:离群细胞 (Outlier) 识别热图。
- 左侧颜色条 (
outlier):TRUE(红色) 代表被判定为离群值的低质量细胞;FALSE(蓝色) 代表保留的优质肿瘤细胞。- 特征:可见离群细胞通常表现为信号杂乱,或与其他肿瘤细胞的拷贝数模式截然不同。
options(repr.plot.width = 12, repr.plot.height = 6, repr.plot.res = 150)
suppressMessages({
ht <- plotHeatmap(tumor_list[['220kb']], label = 'outlier', row_split = 'outlier', n_threads = threads)
ht
})
pdf(file.path(outdir, paste0('plotHeatmap_', samplename, '_220kb_outlier.pdf')), width = 12, height = 6)
ht
dev.off()
tumor_list[['220kb']] <- tumor_list[['220kb']][,colData(tumor_list[['220kb']])$outlier == FALSE]pdf: 2
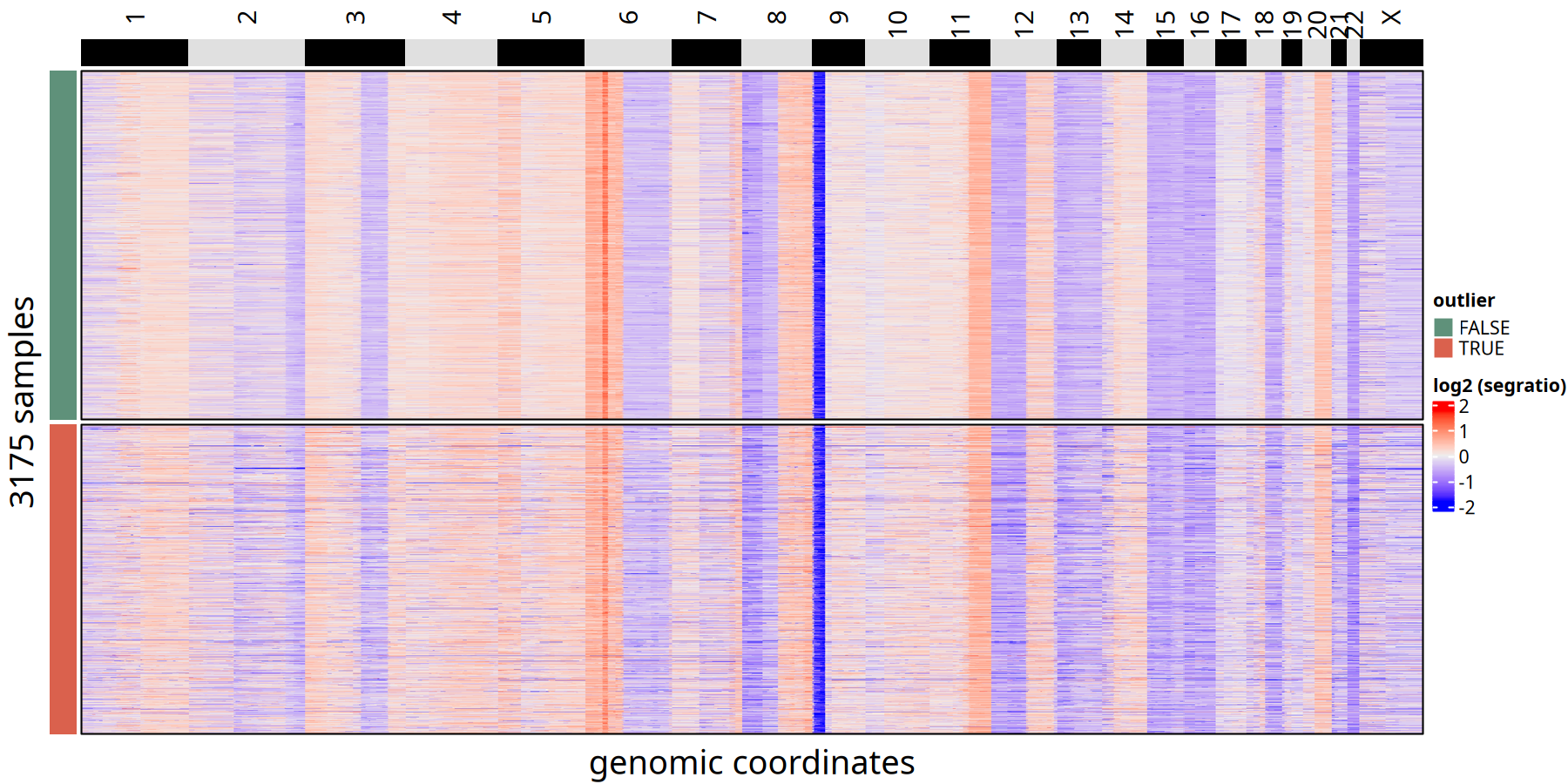
500kb 分辨率下离群细胞与保留细胞的 CNV 热图对比
options(repr.plot.width = 12, repr.plot.height = 6, repr.plot.res = 150)
suppressMessages({
ht <- plotHeatmap(tumor_list[['500kb']], label = 'outlier', row_split = 'outlier', n_threads = threads)
ht
})
pdf(file.path(outdir, paste0('plotHeatmap_', samplename, '_500kb_outlier.pdf')), width = 12, height = 6)
ht
dev.off()
tumor_list[['500kb']] <- tumor_list[['500kb']][,colData(tumor_list[['500kb']])$outlier == FALSE]pdf: 2
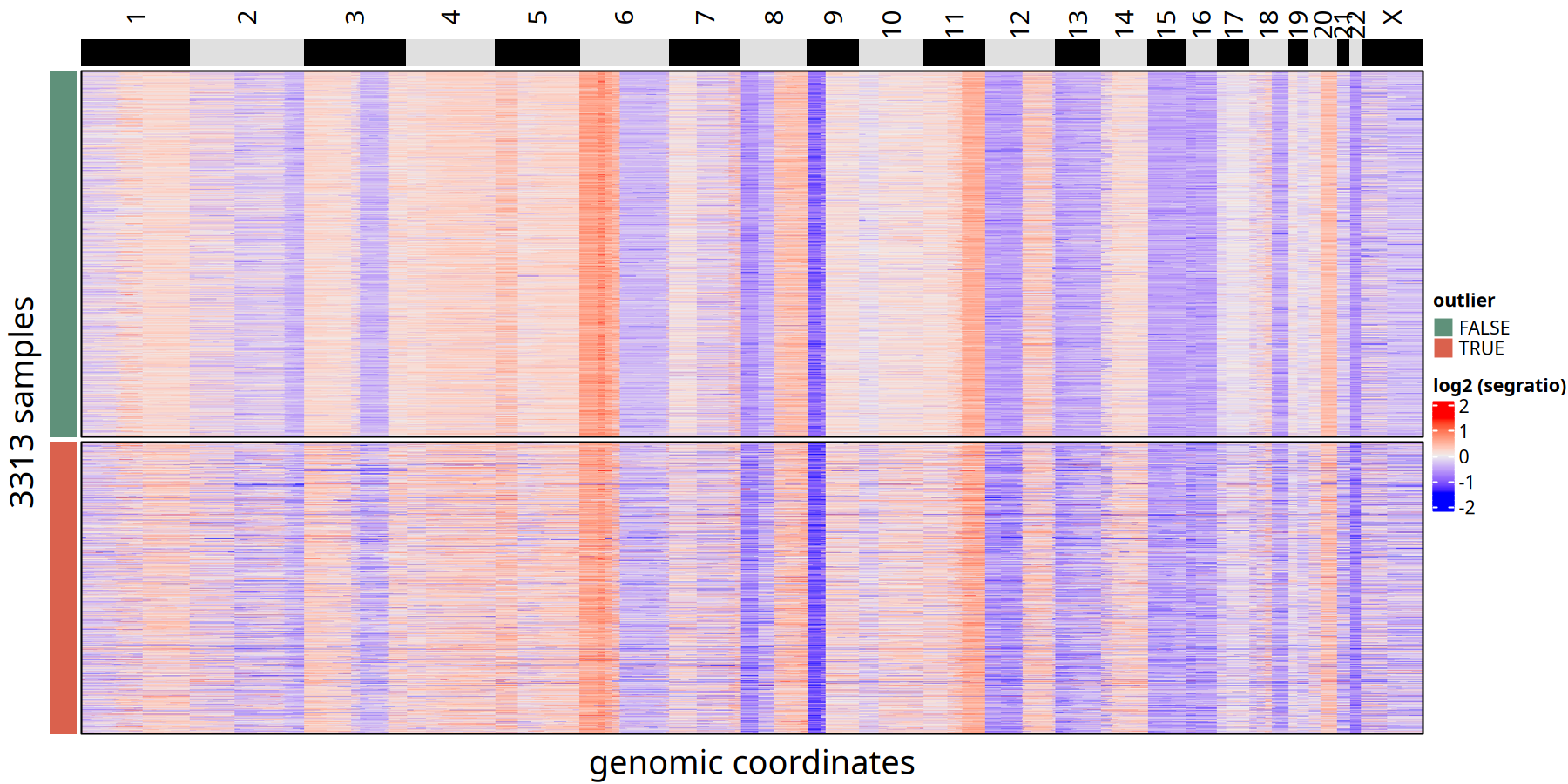
1Mb 分辨率下离群细胞与保留细胞的 CNV 热图对比
options(repr.plot.width = 12, repr.plot.height = 6, repr.plot.res = 150)
suppressMessages({
ht <- plotHeatmap(tumor_list[['1Mb']], label = 'outlier', row_split = 'outlier', n_threads = threads)
ht
})
pdf(file.path(outdir, paste0('plotHeatmap_', samplename, '_1Mbkb_outlier.pdf')), width = 12, height = 6)
ht
dev.off()
tumor_list[['1Mb']] <- tumor_list[['1Mb']][,colData(tumor_list[['1Mb']])$outlier == FALSE]pdf: 2
KNN 平滑处理
由于单细胞测序深度极低(稀疏性高),原始的 Bin Counts 数据往往充满噪声。knnSmooth 函数利用 K-Nearest Neighbors (KNN) 算法,通过整合每个细胞及其最相似的 k 个邻居(默认 k=4)的信息来增强信号,显著降低随机噪声。
- 参数调整:如果数据覆盖度极低,可适当增大
k值,但过大的k值可能会模糊掉亚克隆之间的微小差异。如果您发现后续分群数量过多,可尝试跳过knnSmooth步骤。
if(skipKNNSmooth){
for(i in names(tumor_list)){
suppressMessages({tumor_list[[i]] <- knnSmooth(tumor_list[[i]], k = 4)})
}
saveRDS(tumor_list, file.path(outdir, paste0(samplename, '_copykit_analysis.rds')))
}聚类分析与亚克隆进化分析
降维
为了在二维平面上直观展示肿瘤细胞的克隆结构,我们使用 UMAP (Uniform Manifold Approximation and Projection) 算法对高维的拷贝数数据 (logr 矩阵) 进行降维。
- 含义:在 UMAP 空间中,每个点代表一个细胞。点之间的距离反映了它们全基因组拷贝数图谱的相似程度。聚集在一起的细胞群通常代表具有相同 CNV 特征的 亚克隆 (Subclones)。
for(i in names(tumor_list)){
tumor_list[[i]] <- runUmap(tumor_list[[i]])
}Embedding data with UMAP. Using seed 17
Access reduced dimensions slot with: reducedDim(scCNA, 'umap').
Done.
Using assay: logr
Embedding data with UMAP. Using seed 17
Access reduced dimensions slot with: reducedDim(scCNA, 'umap').
Done.
Using assay: logr
Embedding data with UMAP. Using seed 17
Access reduced dimensions slot with: reducedDim(scCNA, 'umap').
Done.
绝对倍性推断
calcInteger 函数利用 scquantum 算法将 相对拷贝数比率 (Ratios) 转换为 绝对整数拷贝数 (Integer Copy Numbers)。这是因为生物学上的拷贝数必须是整数(如 2, 3, 4)。
- Ploidy Score (倍性评分):该指标衡量了整数拟合的质量。
- 分数接近 0:拟合效果好,细胞的 CNV 状态清晰,容易判断是 2 拷贝还是 3 拷贝。
- 分数较高:说明数据嘈杂,难以准确判断其绝对拷贝数。
- 建议:在后续分析中,可参考 ploidy score 进一步剔除拟合质量极差的细胞。
for(i in names(tumor_list)){
tumor_list[[i]] <- calcInteger(tumor_list[[i]], method = 'scquantum', assay = 'smoothed_bincounts')
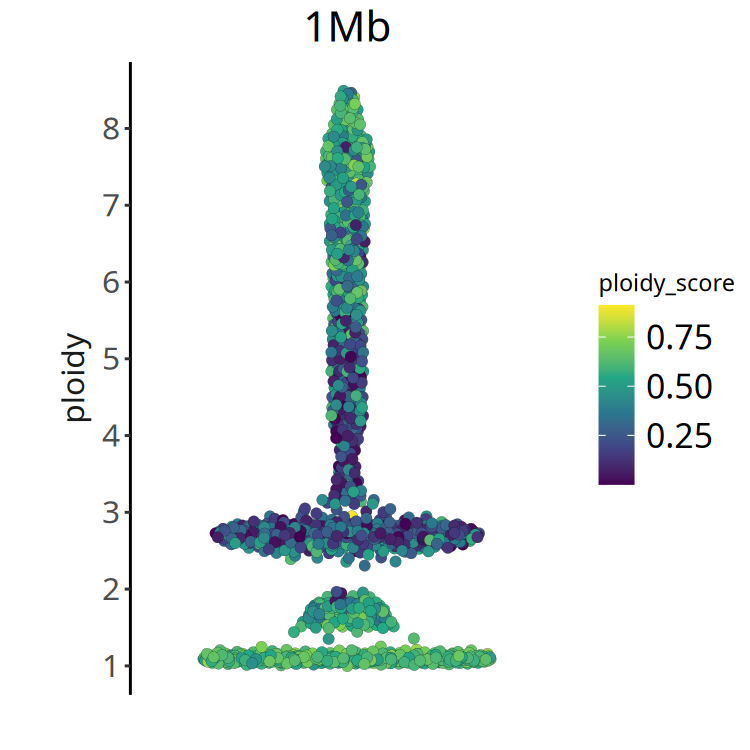
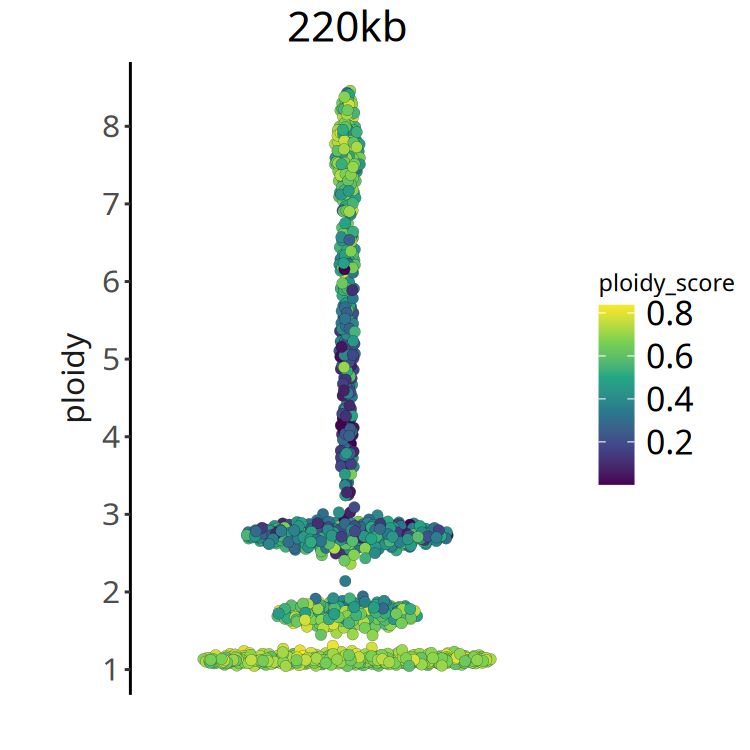
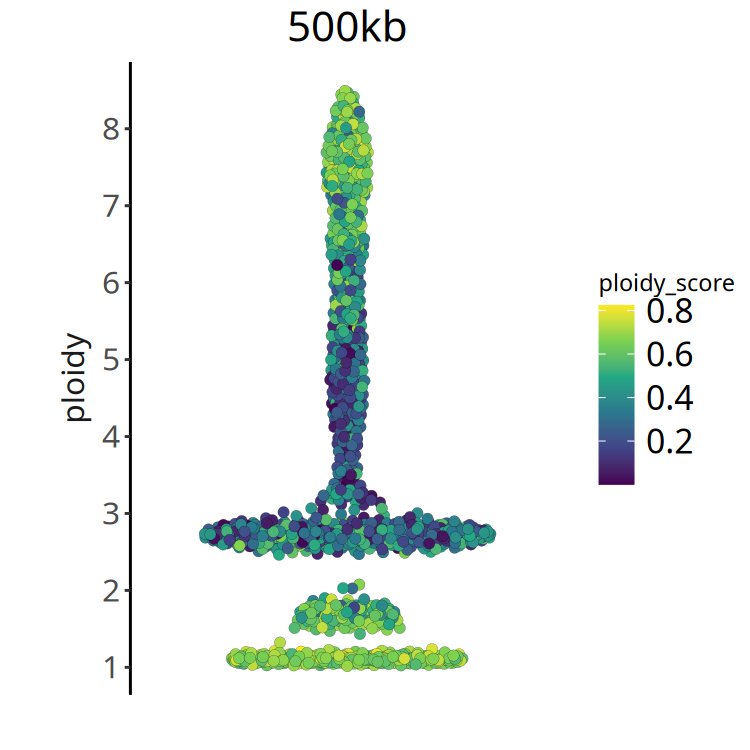
}图注:倍性评分 (Ploidy Score) 分布图。
- X 轴:细胞排序。
- Y 轴:Ploidy Score。
- 颜色:颜色越深(值越小),表示倍性推断越可靠。如果大部分细胞 ploidy score 较大,可能提示样本质量较差或异质性过于复杂。
options(repr.plot.width = 5, repr.plot.height = 5)
for(i in names(tumor_list)){
suppressMessages({
pic <- plotMetrics(
tumor_list[[i]],
metric = 'ploidy',
label = 'ploidy_score') +
ggtitle(i) +
theme(plot.title = element_text(hjust = .5, size = 20))
})
print(pic)
ggsave(file.path(outdir, paste0('ploidy_score_',samplename,'_',i,'.pdf')), pic, width = 5, height = 5)
}
肿瘤亚克隆聚类及克隆进化分析
- 寻找最佳 K 值 (
findSuggestedK):通过计算不同 K 值下的 Jaccard 相似度,自动推荐最适合的聚类邻居数 (Nearest Neighbors)。 - 执行聚类 (
findClusters):基于 UMAP 的坐标进行无监督聚类,识别不同的肿瘤亚克隆 (Subclones)。默认采用 HDBSCAN 算法。相比于传统的 Louvain/Leiden,HDBSCAN 能更灵敏地识别出噪声点(标记为c0)。 - 构建共识图谱 (Consensus Profiles):单细胞数据不可避免地带有随机噪声。为了准确推断肿瘤的进化历史,我们不再关注单个细胞的微小波动,而是通过
calcConsensus计算每个亚克隆的共识拷贝数图谱 (Consensus Profile)。该函数取该亚克隆内所有细胞在每个 Bin 上的中位数 (Median),构建出一个代表该克隆特征的“虚拟细胞”。这极大地提高了信噪比。 - 构建亚克隆进化树:
runConsensusPhylo函数基于各个亚克隆的共识图谱,计算它们之间的曼哈顿距离 (Manhattan Distance),并构建系统发育树。
关于离群簇 'c0'
HDBSCAN 算法可能会将一部分细胞归类为
c0簇。这些细胞通常因为以下原因无法归入任何主要克隆:
- 数据噪声:测序质量低导致拷贝数特征模糊。
- 罕见克隆:细胞数量太少,未达到独立成簇的最小阈值。
- 双细胞/降解:由技术误差导致的异常信号。
处理建议:在分析肿瘤主要的进化结构之前,通常建议移除
c0簇,以免干扰进化树的构建。
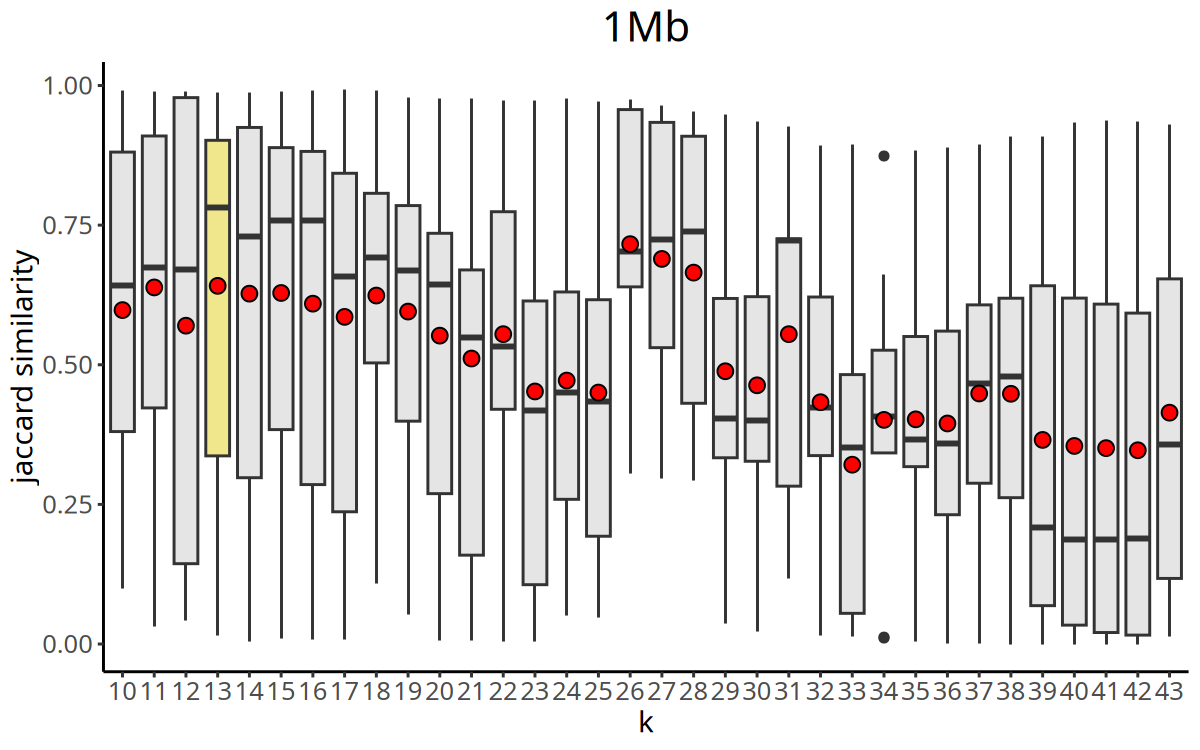
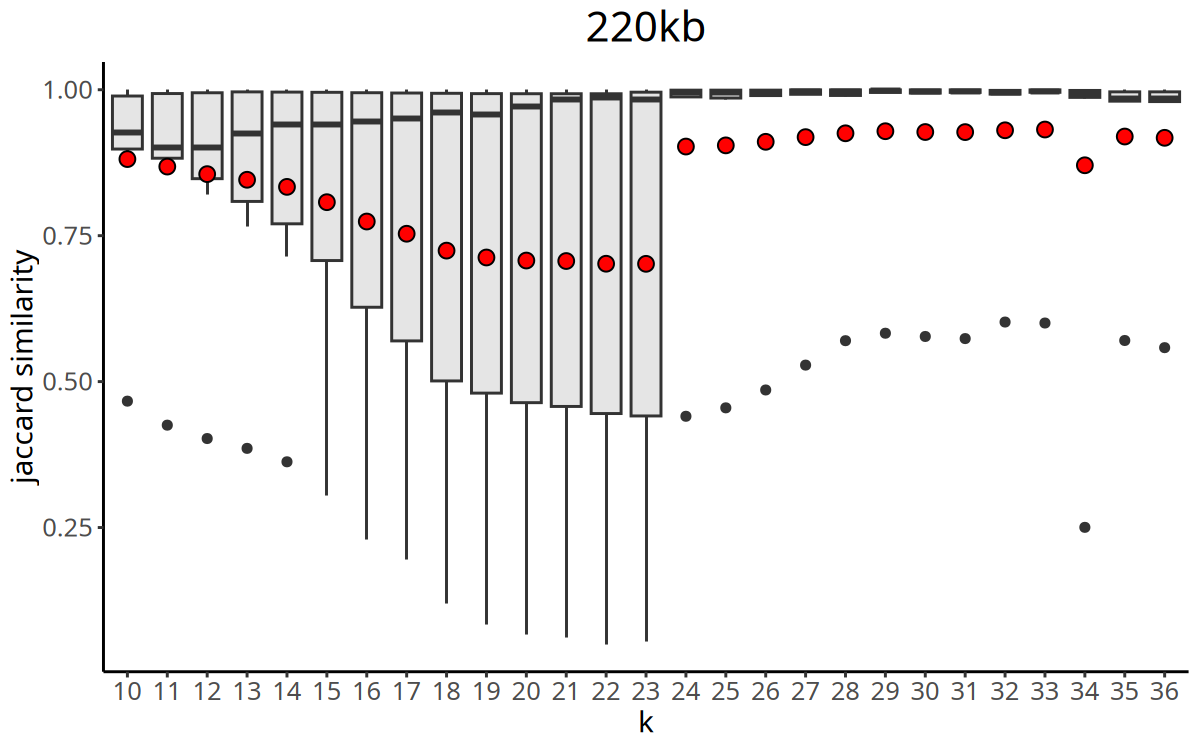
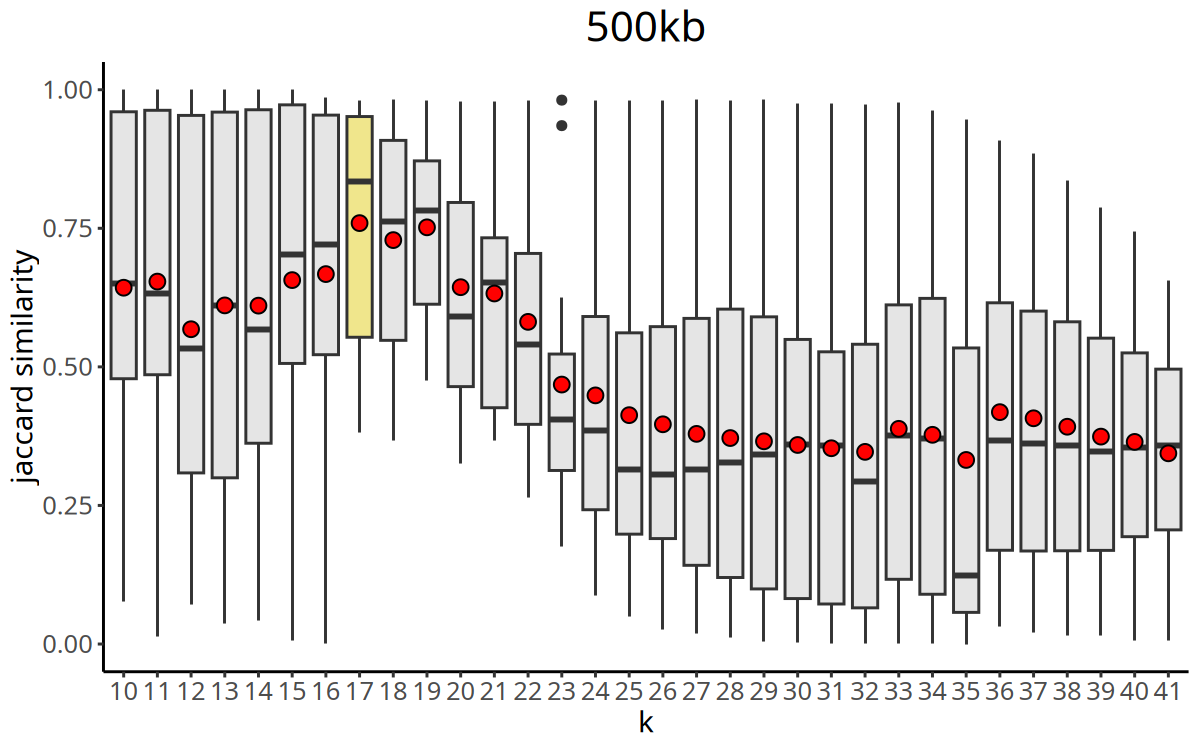
图注:Jaccard 相似度指数随 K 值变化的曲线。
- X 轴:K 值(最近邻居数)。
- Y 轴:Jaccard Index(聚类稳定性指标)。
CopyKit 会自动选择 Jaccard 相似度最大的 K 值作为后续
findClusters的参数。
options(repr.plot.width = 8, repr.plot.height = 5)
for(i in names(tumor_list)){
suppressMessages({tumor_list[[i]] <- findSuggestedK(tumor_list[[i]])})
pic <- plotSuggestedK(tumor_list[[i]]) +
ggtitle(i) +
theme(plot.title = element_text(hjust = .5, size = 20))
print(pic)
ggsave(file.path(outdir, paste0('SuggestedK_',samplename,'_',i,'.pdf')), pic, width = 8, height = 5)
}
for(i in names(tumor_list)){
tumor_list[[i]] <- findClusters(tumor_list[[i]])
}Finding clusters, using method: hdbscan
Found 495 outliers cells (group 'c0')
Found 15 subclones.
Done.
Using suggested k_subclones = 29
Finding clusters, using method: hdbscan
Found 2 outliers cells (group 'c0')
Found 5 subclones.
Done.
Using suggested k_subclones = 17
Finding clusters, using method: hdbscan
Found 438 outliers cells (group 'c0')
Found 11 subclones.
Done.
220kb 分辨率下肿瘤亚克隆分析结果
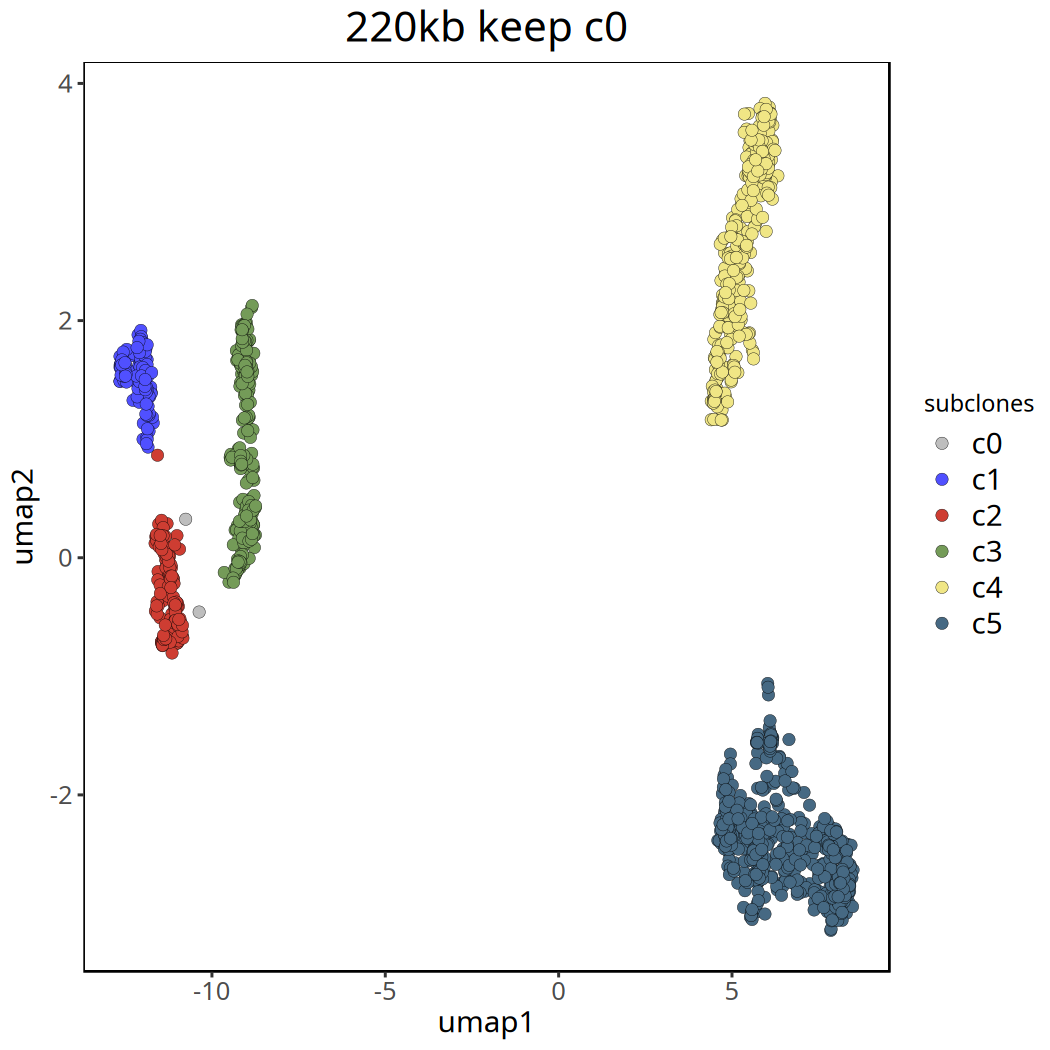
以下展示了去除 c0 群前后的 UMAP 聚类图。
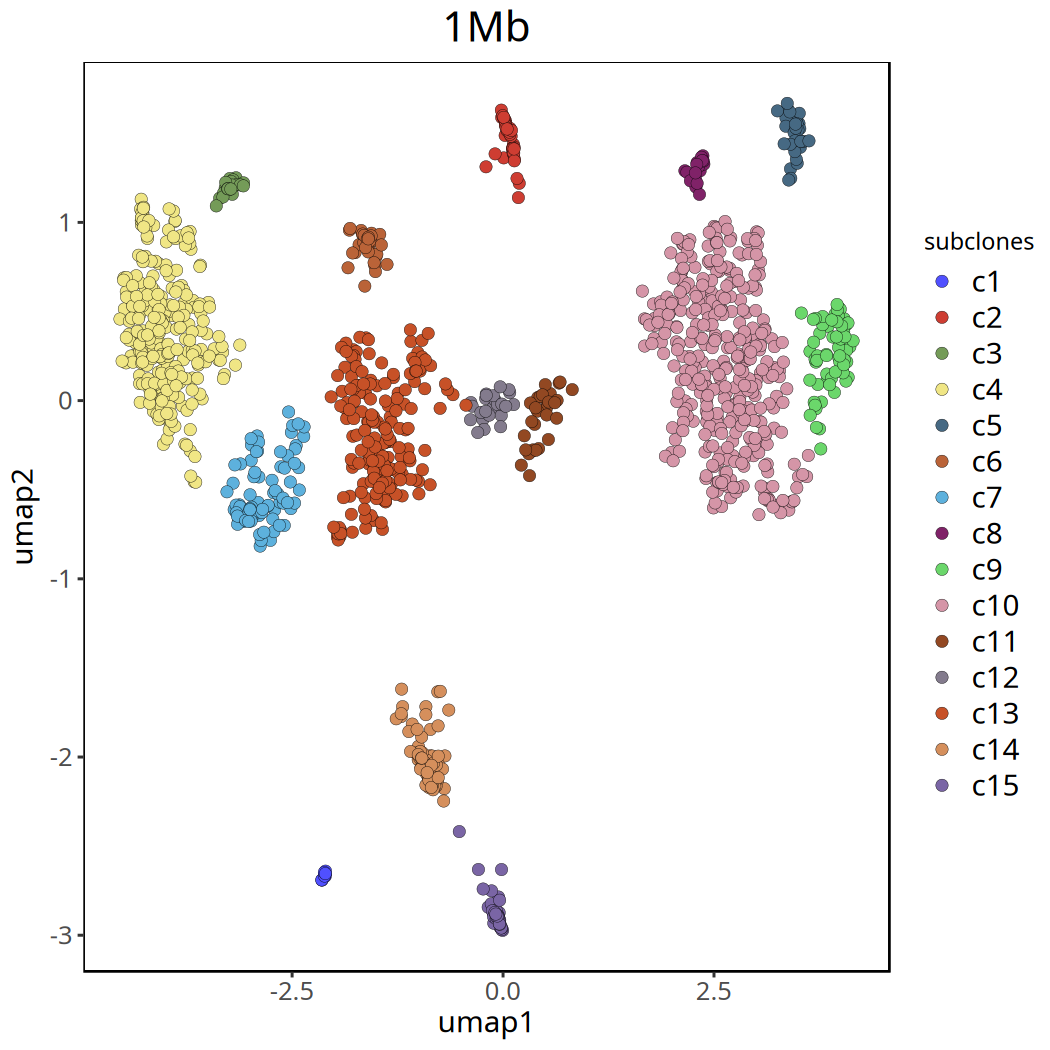
图注:移除
c0后的 UMAP 聚类图。不同颜色清晰地展示了肿瘤内部的主要亚克隆结构。
options(repr.plot.width = 7, repr.plot.height = 7)
pic <- plotUmap(tumor_list[['220kb']], label = 'subclones') +
ggtitle('220kb keep c0') +
theme(plot.title = element_text(hjust = .5, size = 20))
pic
ggsave(file.path(outdir, paste0('plotUmap',samplename,'_',i,'_keep_c0.pdf')), pic, width = 7, height = 7)Coloring by: subclones.
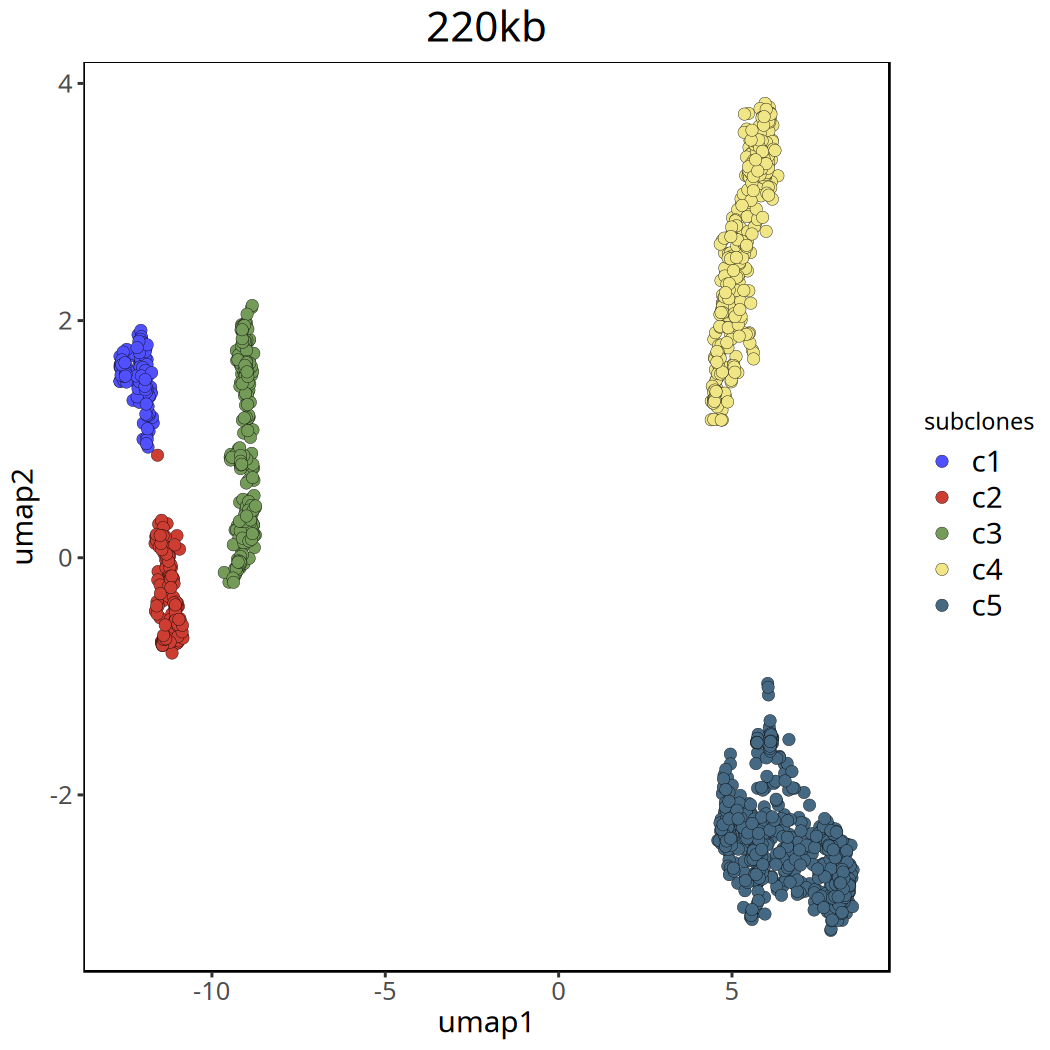
tumor_list[['220kb']] <- tumor_list[['220kb']][,colData(tumor_list[['220kb']])$subclones != 'c0']
pic <- plotUmap(tumor_list[['220kb']], label = 'subclones') +
ggtitle('220kb') +
theme(plot.title = element_text(hjust = .5, size = 20))
pic
ggsave(file.path(outdir, paste0('plotUmap',samplename,'_',i,'_remove_c0.pdf')), pic, width = 7, height = 5)Coloring by: subclones.
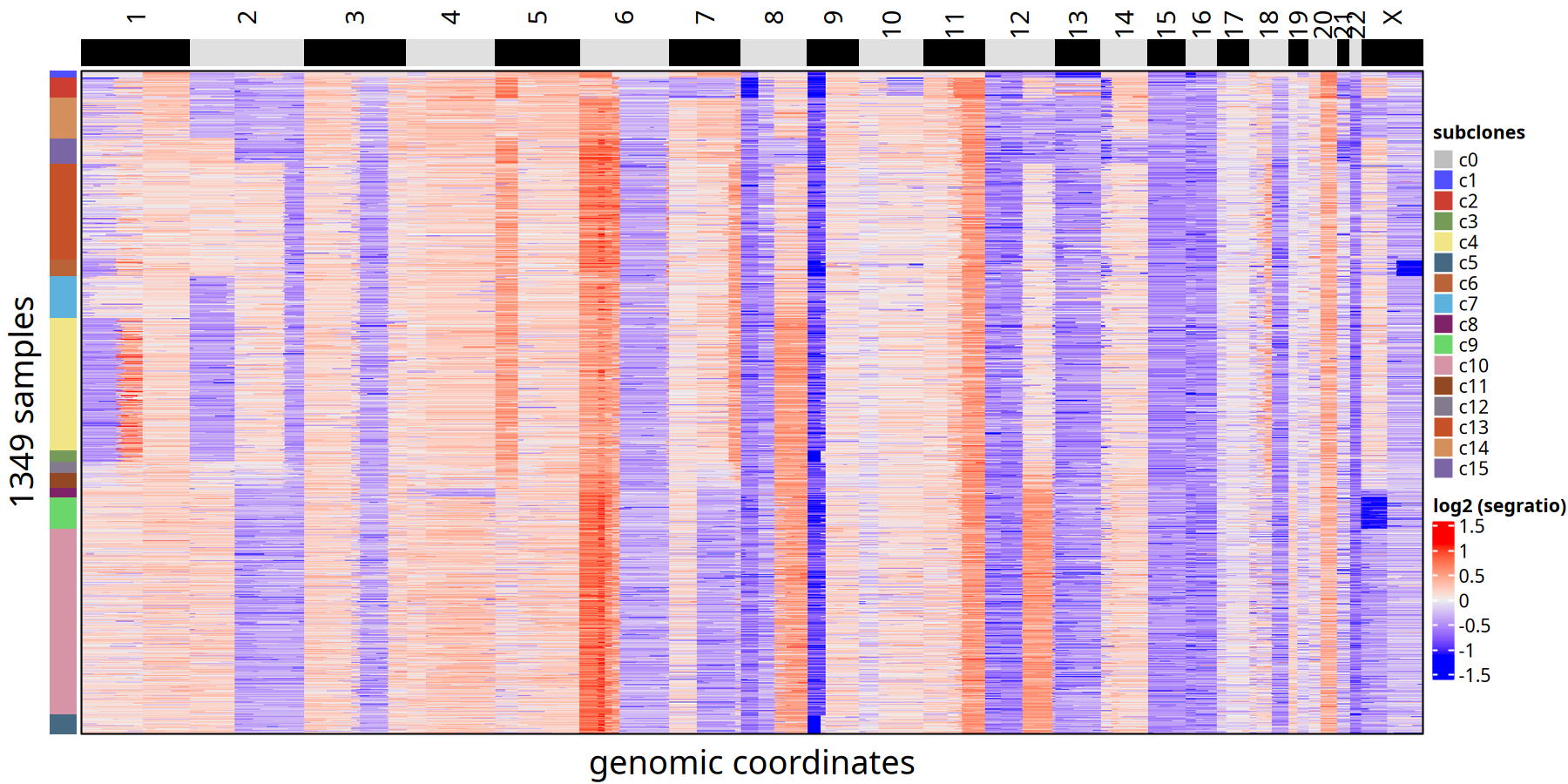
220kb 分辨率下肿瘤亚克隆 CNV 热图以及亚克隆进化树
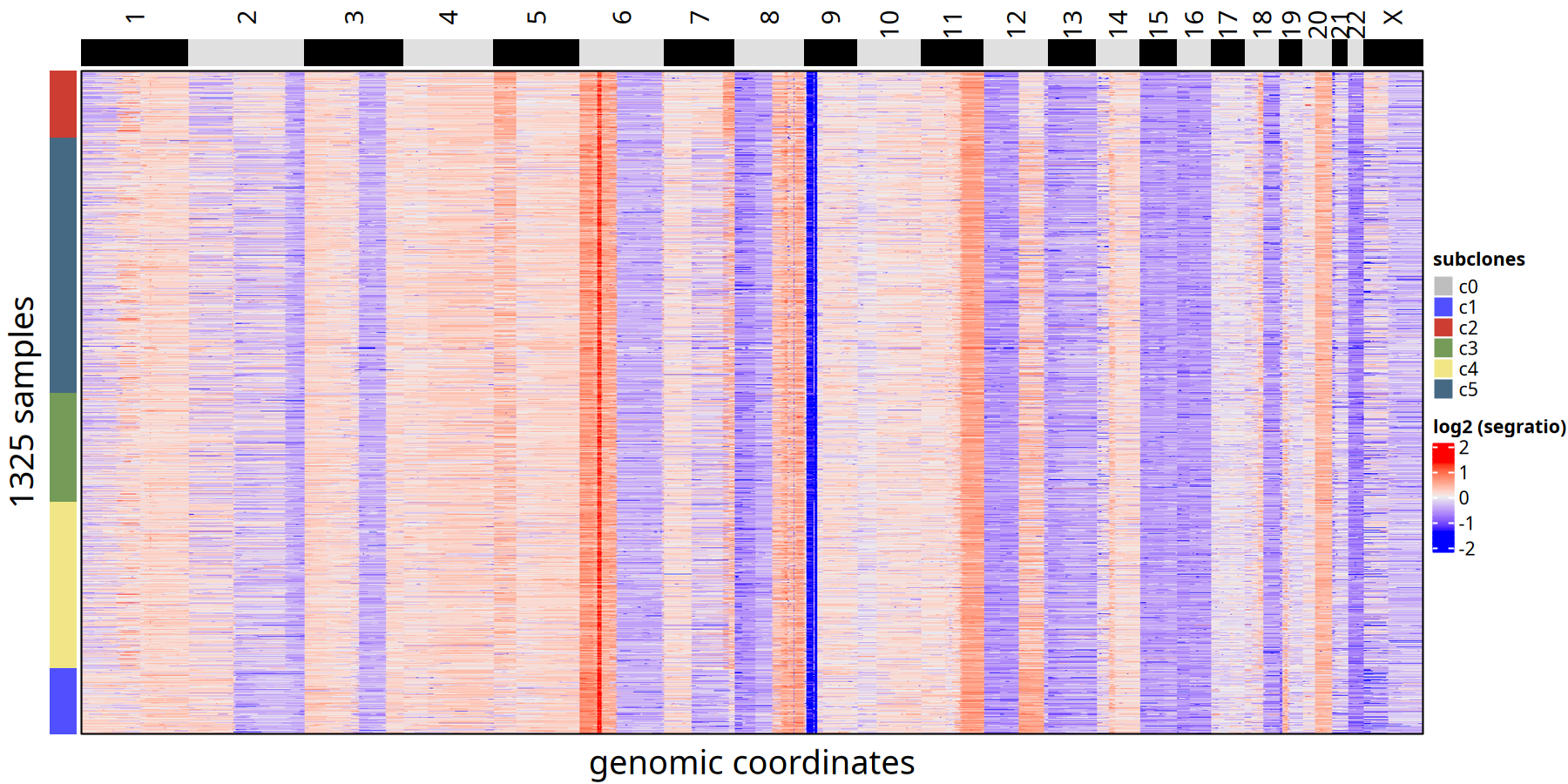
图注:亚克隆共识热图
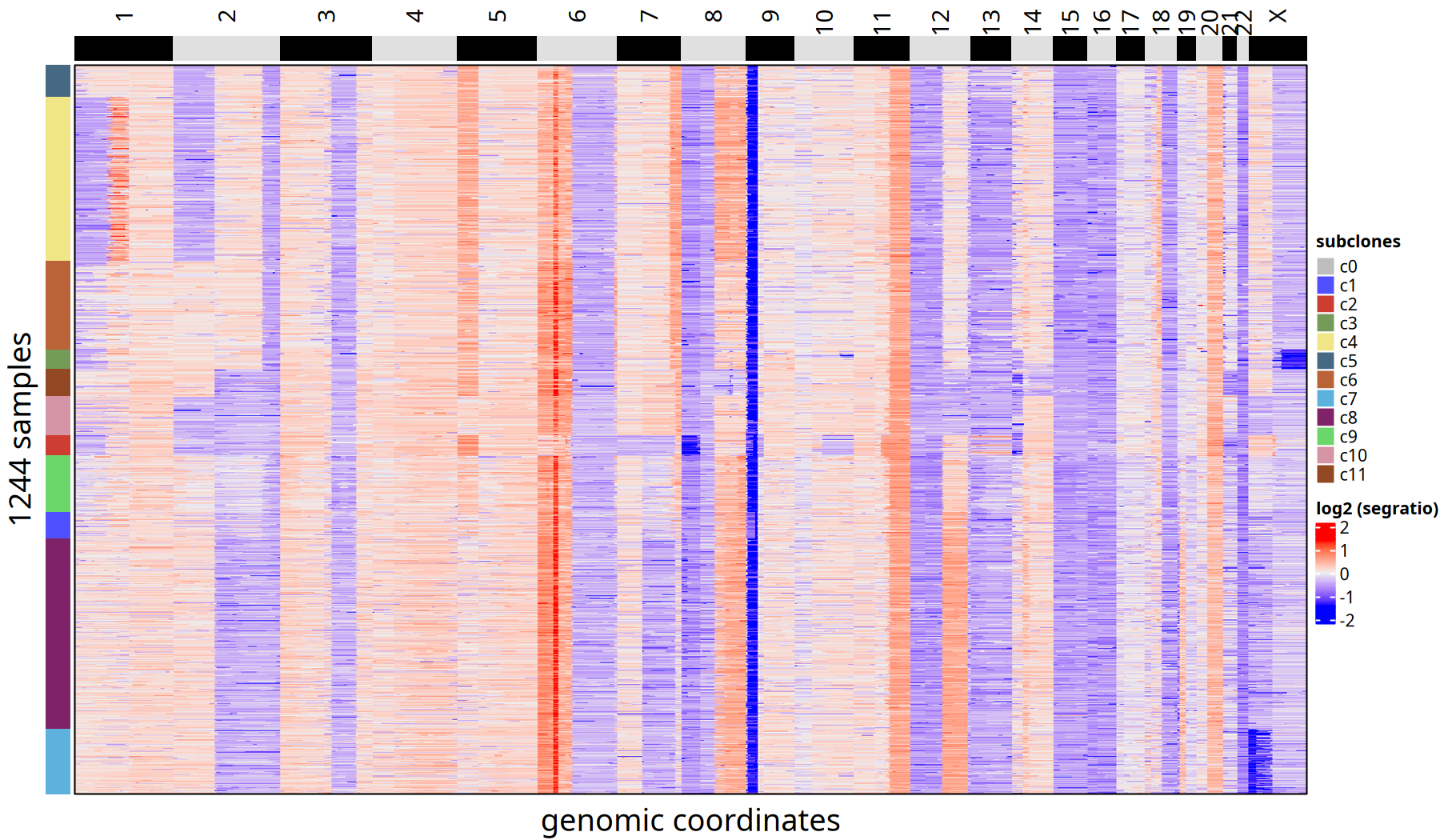
- 热图:每一行代表一个细胞,按进化树顺序排列。颜色代表 Log2 Segment Ratio。
- 左侧注释:标注了细胞所属的亚克隆编号。
options(repr.plot.width = 12, repr.plot.height = 6, repr.plot.res = 150)
tumor_list[['220kb']] <- calcConsensus(tumor_list[['220kb']])
tumor_list[['220kb']] <- runConsensusPhylo(tumor_list[['220kb']])
suppressMessages({
ht <- plotHeatmap(
tumor_list[['220kb']],
label = 'subclones',
order_cells = 'consensus_tree'
)
})
ht
pdf(
file.path(outdir, paste0('plotHeatmap_consensus_tree_',samplename,'_',i,'.pdf')),
width = 16,
height = 6)
ht
dev.off()pdf: 2
图注:亚克隆系统发育树 (Phylogenetic Tree)。
- 枝长:代表曼哈顿距离,即亚克隆之间基因组 CNV 图谱的差异大小。
- 结构:展示了肿瘤如何从根部演化出不同的分支(亚克隆)。
suppressWarnings({
pic <- plotPhylo(tumor_list[['220kb']], label = 'subclones', consensus = TRUE)
})
pic
ggsave(file.path(outdir, paste0('plotPhylo_consensus_',samplename,'_220kb','.pdf')), pic, width = 7, height = 5)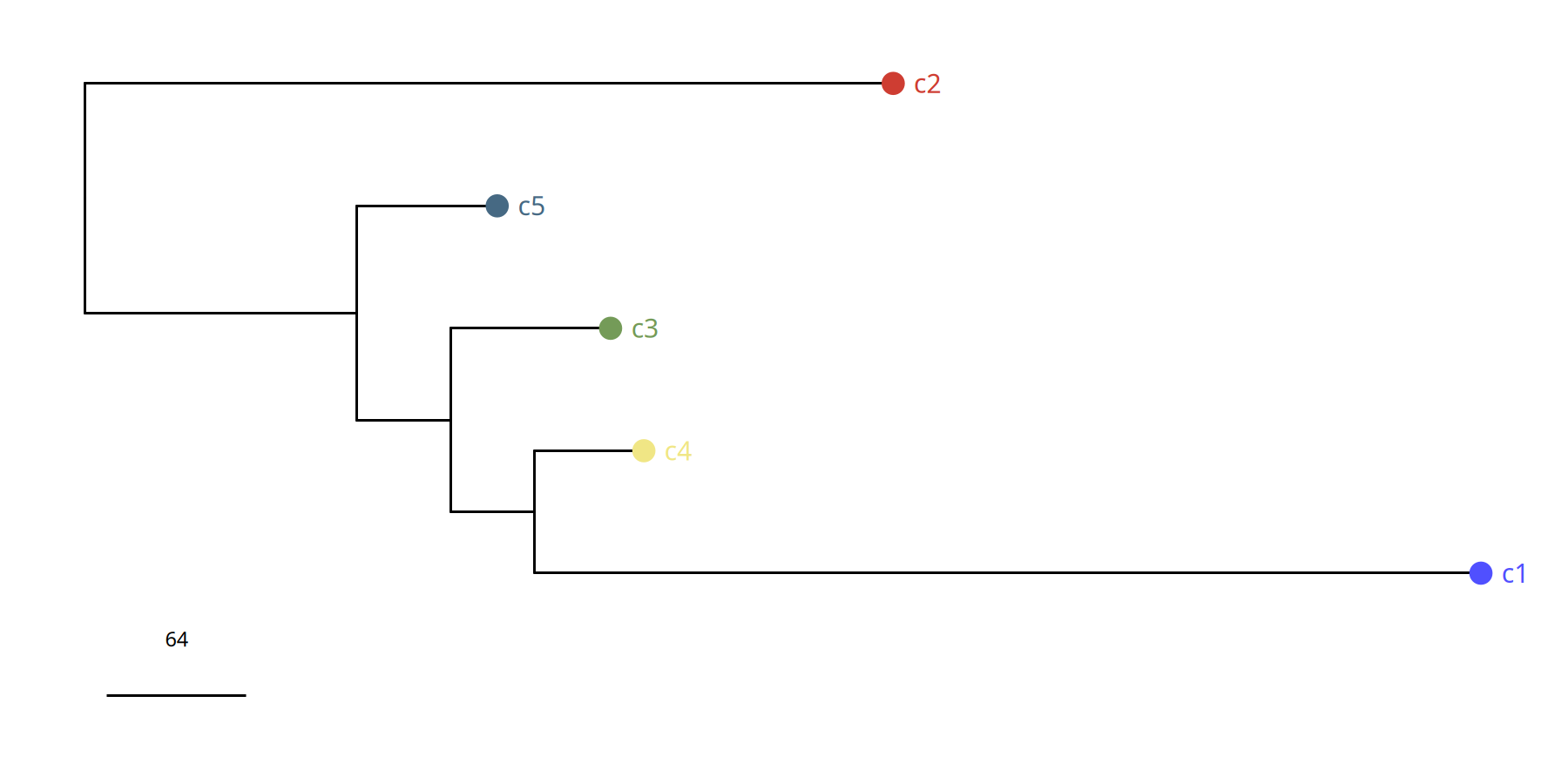
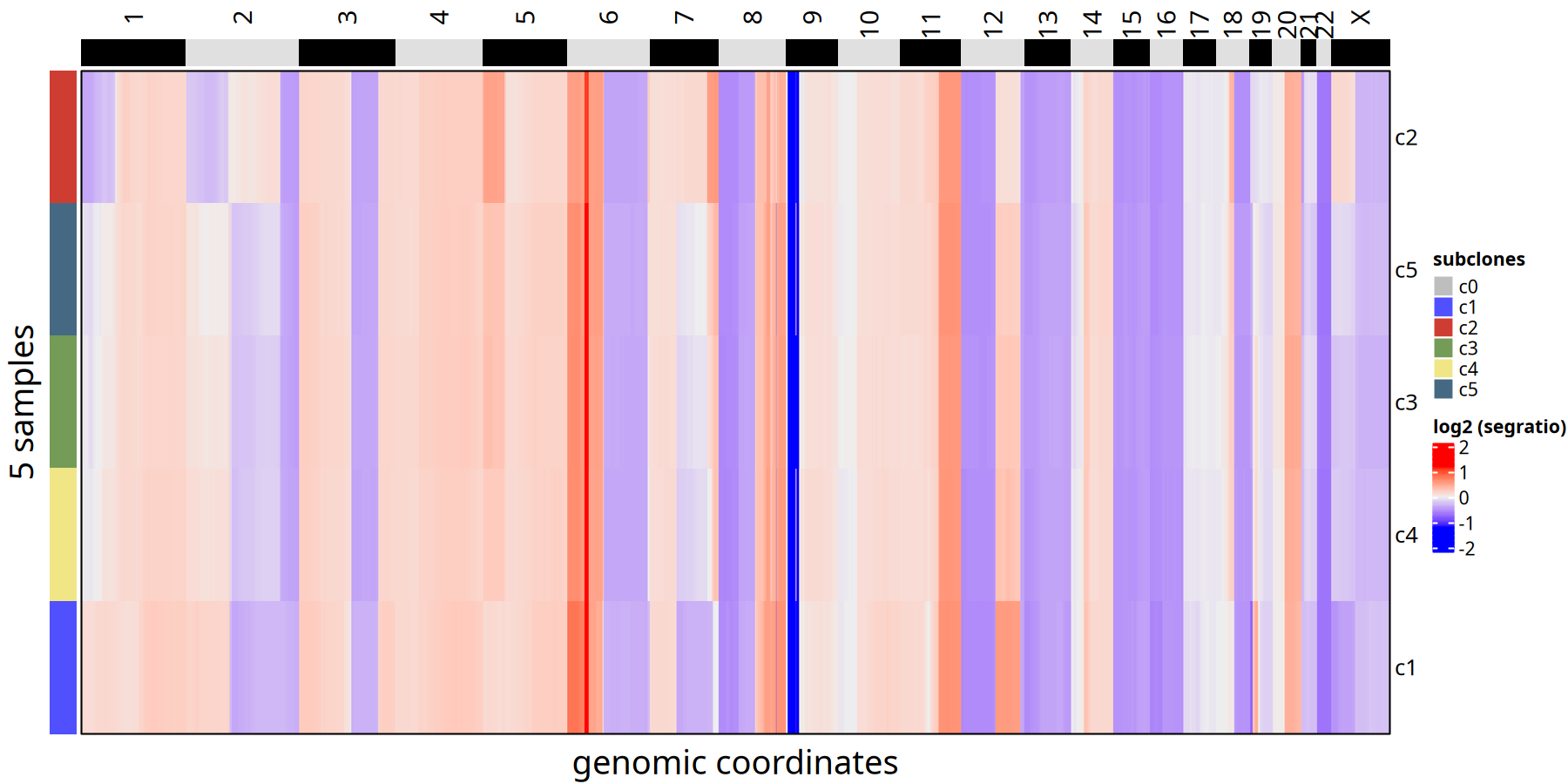
以下热图和折线图是从“Bulk”水平(即亚克隆共识水平)上观察不同亚克隆之间 CNV 的差异。
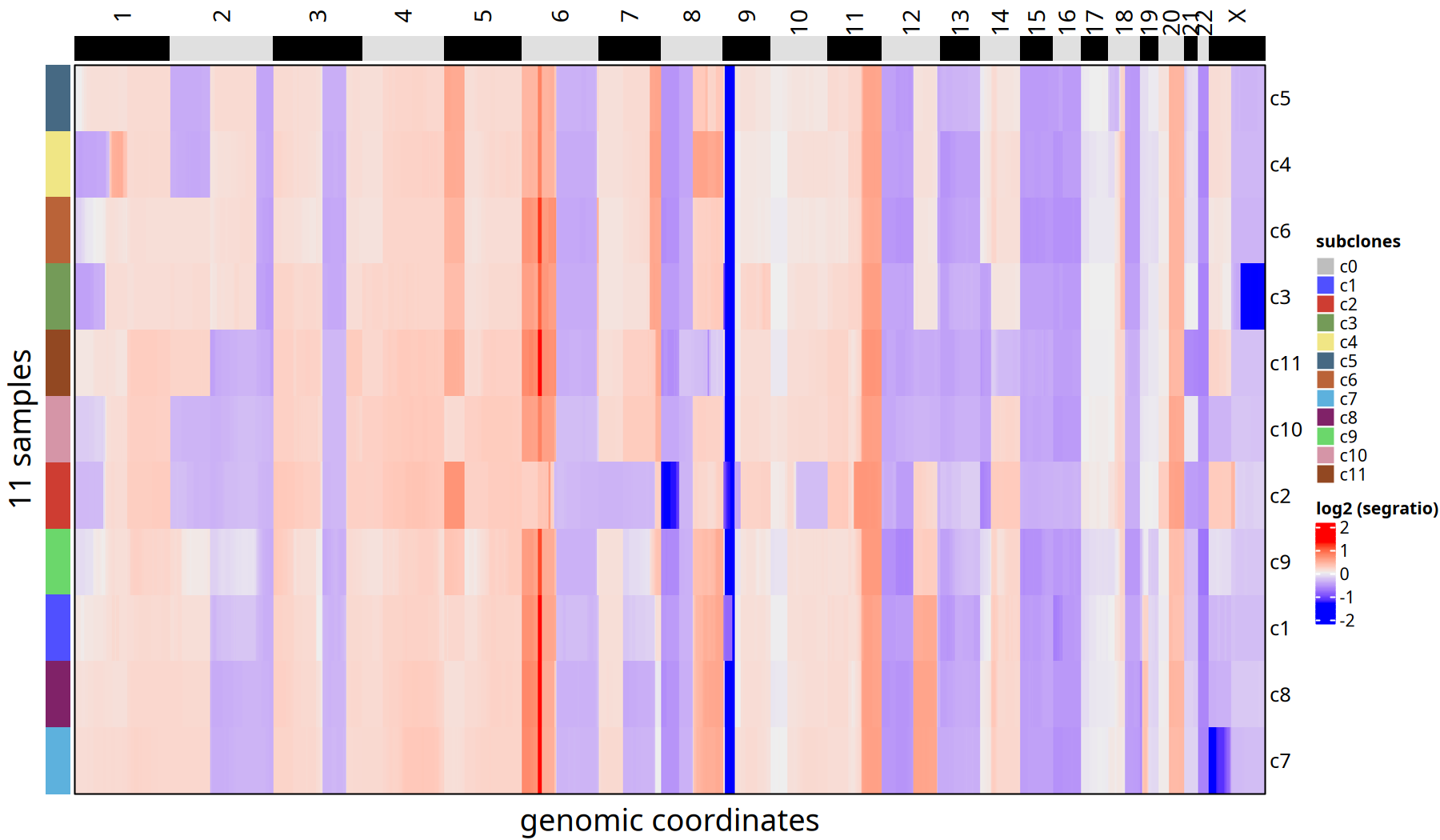
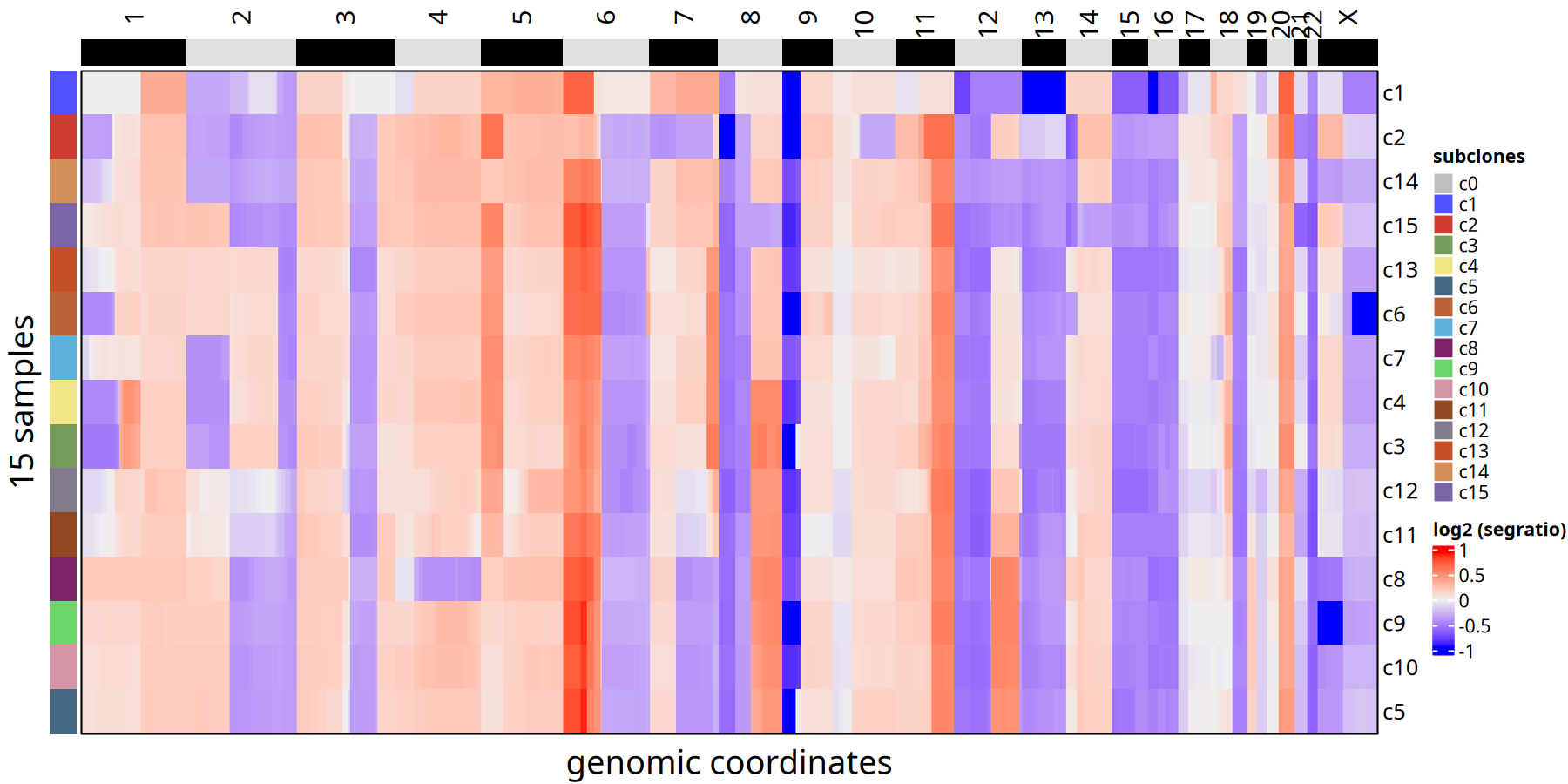
图注:亚克隆共识热图 (Consensus Heatmap)。
与单细胞热图不同,这里每一行仅代表一个亚克隆(而非单个细胞)。这消除了单细胞噪声,清晰展示了每个亚克隆的核心 CNV 事件(如某染色体臂的整体扩增或缺失)。
tumor_list[['220kb']] <- calcConsensus(tumor_list[['220kb']] )
suppressMessages({
ht <- plotHeatmap(tumor_list[['220kb']],consensus = TRUE,label = 'subclones')
})
ht
pdf(
file.path(outdir, paste0('plotHeatmap_consensus_tree_',samplename,'_220kb','.pdf')),
width = 16,
height = 6)
ht
dev.off()pdf: 2
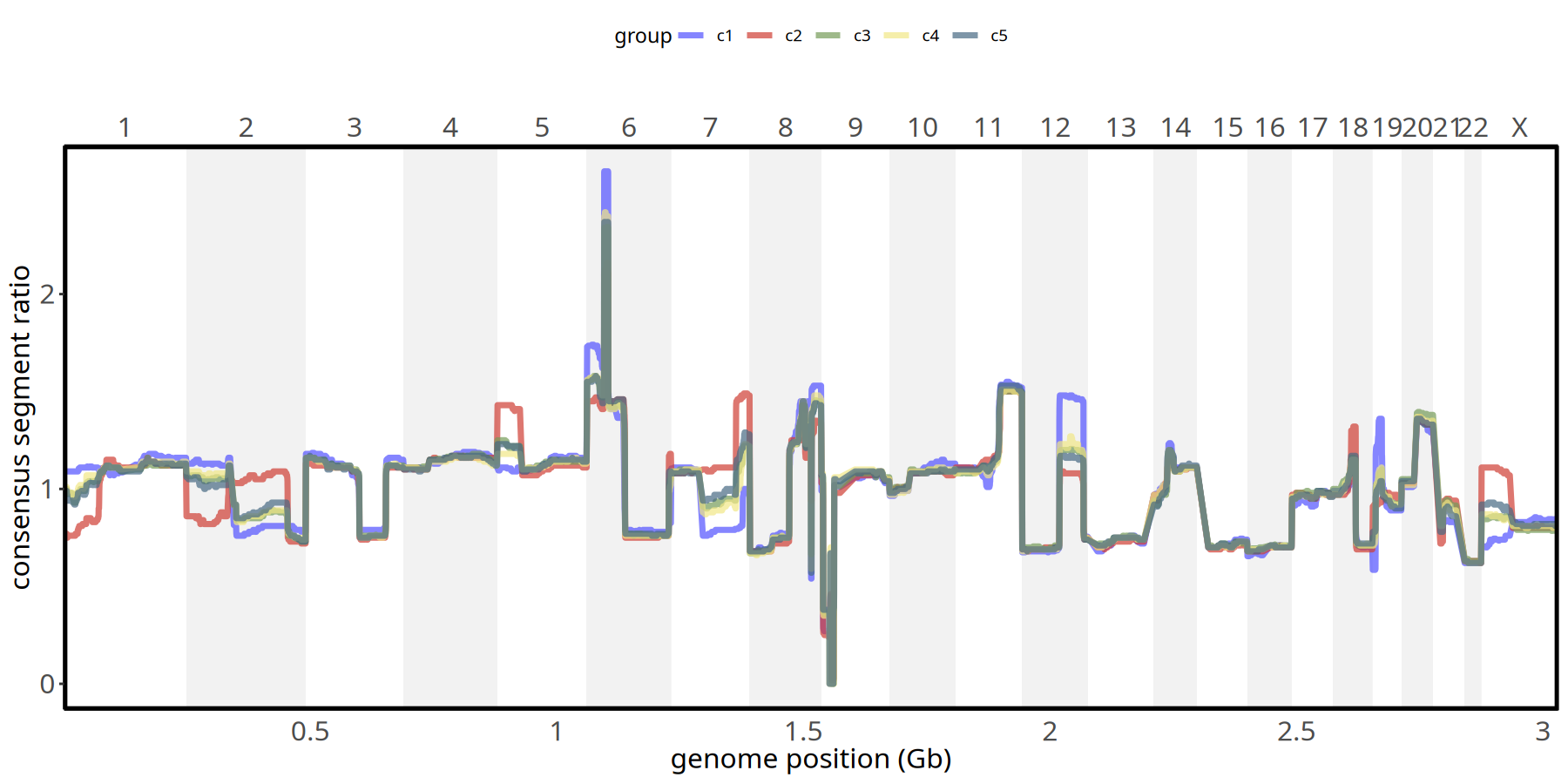
图注:亚克隆拷贝数分布曲线图。
- X 轴:基因组染色体位置 (chr1 - chr22)。
- Y 轴:Consensus Segment Ratio (共识片段比率, 取得是 subclone 内部每个 segment 的中位值)。
- 含义:不同颜色的线条代表不同亚克隆。线条在基线 (1.0) 以上的隆起代表扩增,凹陷代表缺失。通过此图可精确比较不同亚克隆在特定位点的差异。
pic <- plotConsensusLine_static(tumor_list[['220kb']],levels(colData(tumor_list[['220kb']])$subclones) ) +
scale_color_manual(values = subclones_pal())
pic
ggsave(file.path(outdir, paste0('plot_ConsensusLine_',samplename,'_220kb','.pdf')), pic, width = 7, height = 5)“The \`size\` argument of \`element_rect()\` is deprecated as of ggplot2 3.4.0.
ℹ Please use the \`linewidth\` argument instead.”
Warning message:
“Using \`size\` aesthetic for lines was deprecated in ggplot2 3.4.0.
ℹ Please use \`linewidth\` instead.”
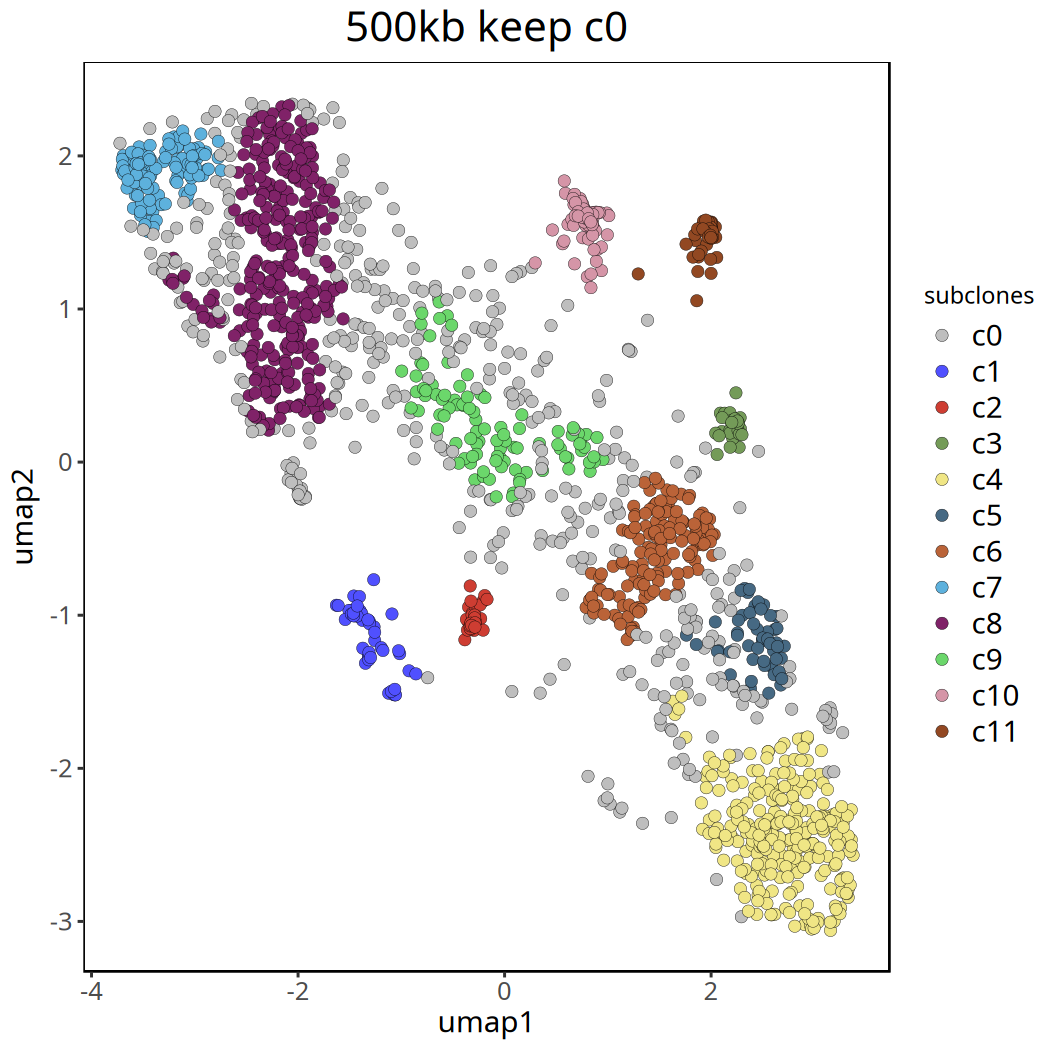
500kb 分辨率下肿瘤亚克隆分析结果
以下展示了去除 c0 群前后的 UMAP 聚类图。
options(repr.plot.width = 7, repr.plot.height = 7)
pic <- plotUmap(tumor_list[['500kb']], label = 'subclones') +
ggtitle('500kb keep c0') +
theme(plot.title = ggplot2::element_text(hjust = .5, size = 20))
pic
ggsave(file.path(outdir, paste0('plotUmap',samplename,'_1Mb','_keep_c0.pdf')), pic, width = 7, height = 5)Coloring by: subclones.
tumor_list[['500kb']] <- tumor_list[['500kb']][,colData(tumor_list[['500kb']])$subclones != 'c0']
pic <- plotUmap(tumor_list[['500kb']], label = 'subclones') +
ggtitle('500kb') +
theme(plot.title = element_text(hjust = .5, size = 20))
pic
ggsave(file.path(outdir, paste0('plotUmap',samplename,'_500kb','_remove_c0.pdf')), pic, width = 7, height = 5)Coloring by: subclones.
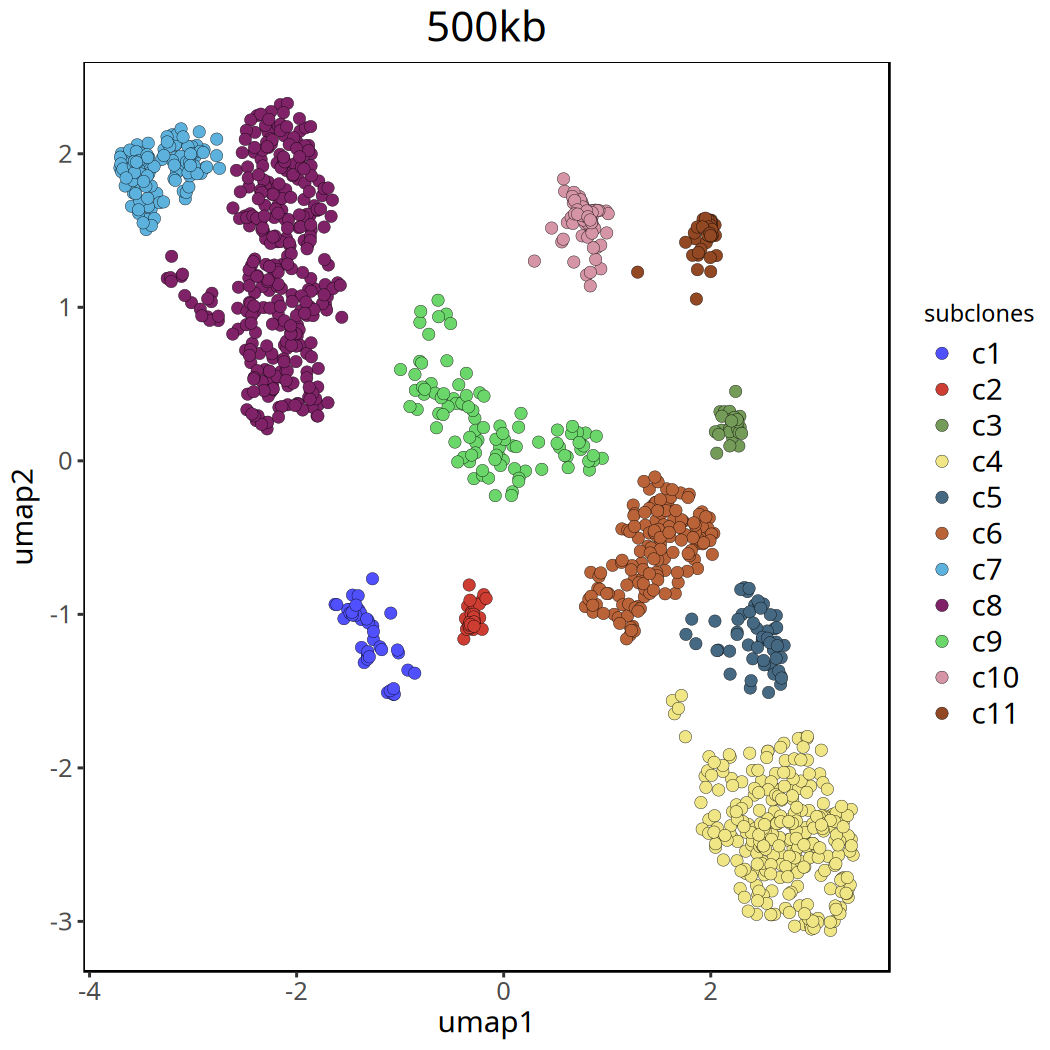
500kb 分辨率下肿瘤亚克隆 CNV 热图以及亚克隆进化树
options(repr.plot.width = 12, repr.plot.height = 7, repr.plot.res = 150)
tumor_list[['500kb']] <- calcConsensus(tumor_list[['500kb']])
tumor_list[['500kb']] <- runConsensusPhylo(tumor_list[['500kb']])
suppressMessages({
ht <- plotHeatmap(
tumor_list[['500kb']],
label = 'subclones',
order_cells = 'consensus_tree'
)})
ht
pdf(
file.path(outdir, paste0('plotHeatmap_consensus_tree_',samplename,'_550kb','.pdf')),
width = 16,
height = 6)
ht
dev.off()pdf: 2
suppressWarnings({
pic <- plotPhylo(tumor_list[['500kb']], label = 'subclones', consensus = TRUE)
})
pic
ggsave(file.path(outdir, paste0('plotPhylo_consensus_',samplename,'_500kb','.pdf')), pic, width = 7, height = 7)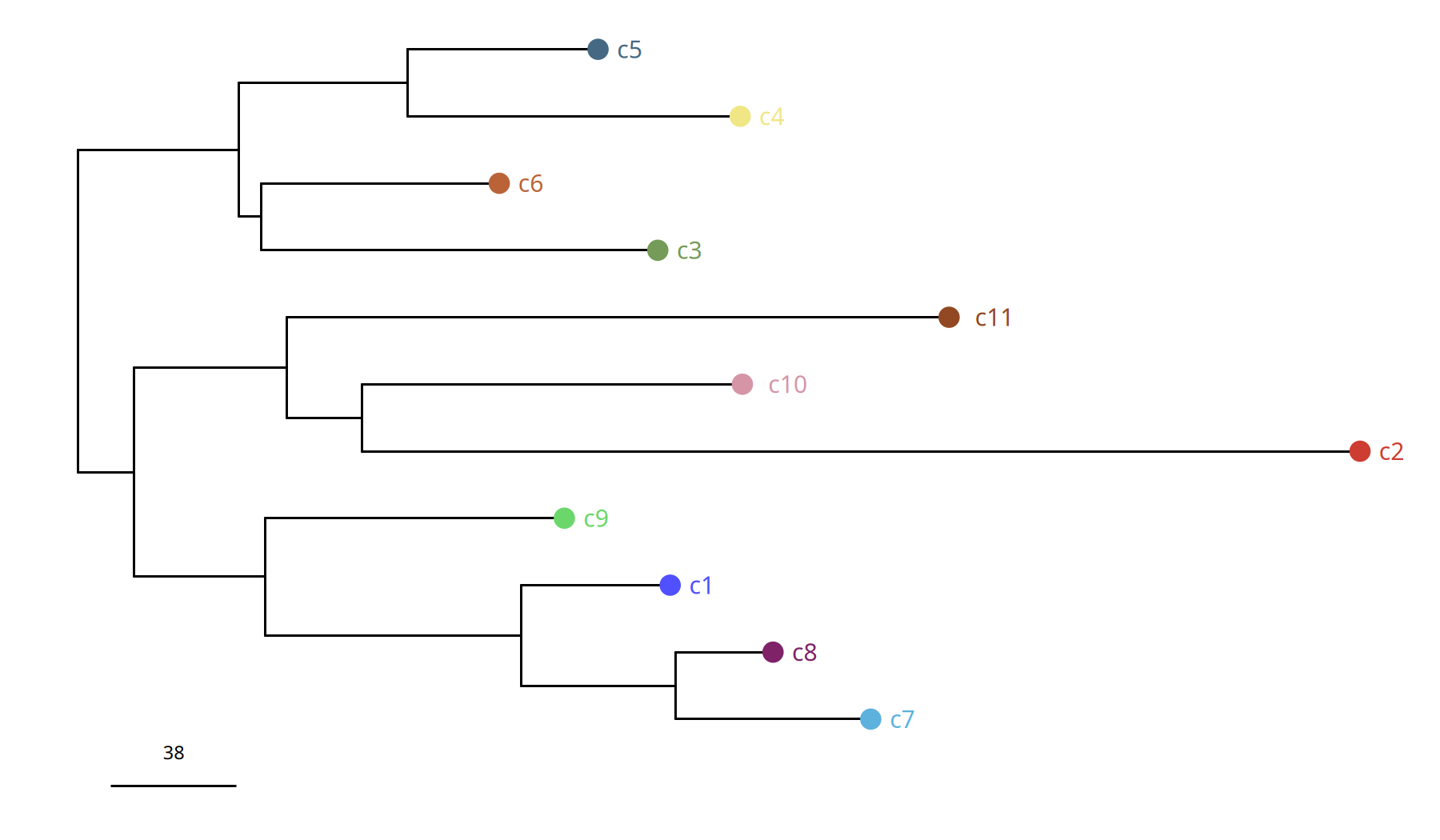
以下热图和折线图是从“Bulk”水平上观察不同亚克隆之间 CNV 的差异。
tumor_list[['500kb']] <- calcConsensus(tumor_list[['500kb']] )
suppressMessages({
ht <- plotHeatmap(tumor_list[['500kb']],consensus = TRUE,label = 'subclones')
})
ht
pdf(
file.path(outdir, paste0('plotHeatmap_consensus_',samplename,'_550kb','.pdf')),
width = 16,
height = 6)
ht
dev.off()pdf: 2
pic <- plotConsensusLine_static(tumor_list[['500kb']],levels(colData(tumor_list[['500kb']])$subclones) ) +
scale_color_manual(values = subclones_pal())
pic
ggsave(file.path(outdir, paste0('plot_ConsensusLine_',samplename,'_500kb','.pdf')), pic, width = 7, height = 5)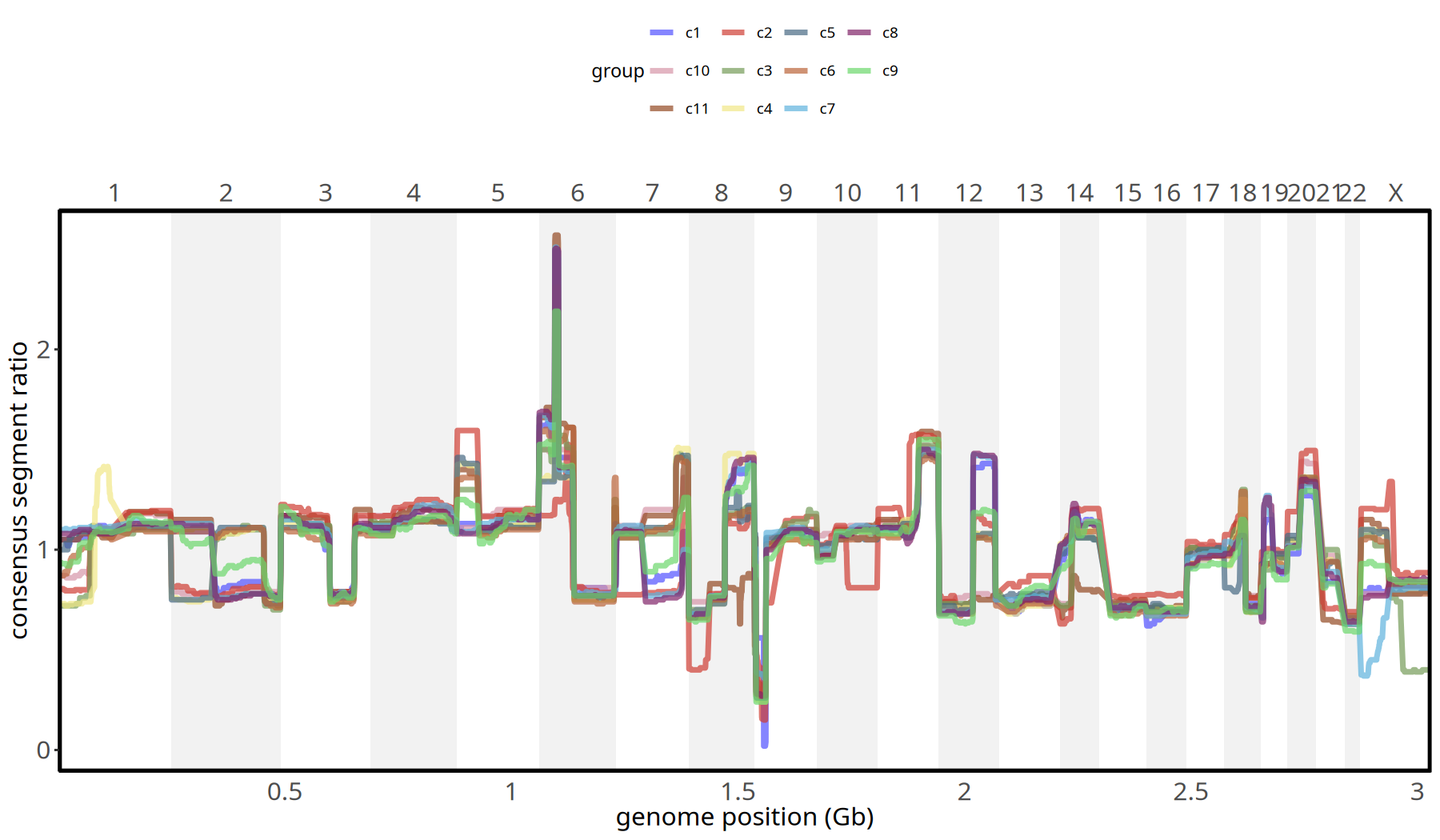
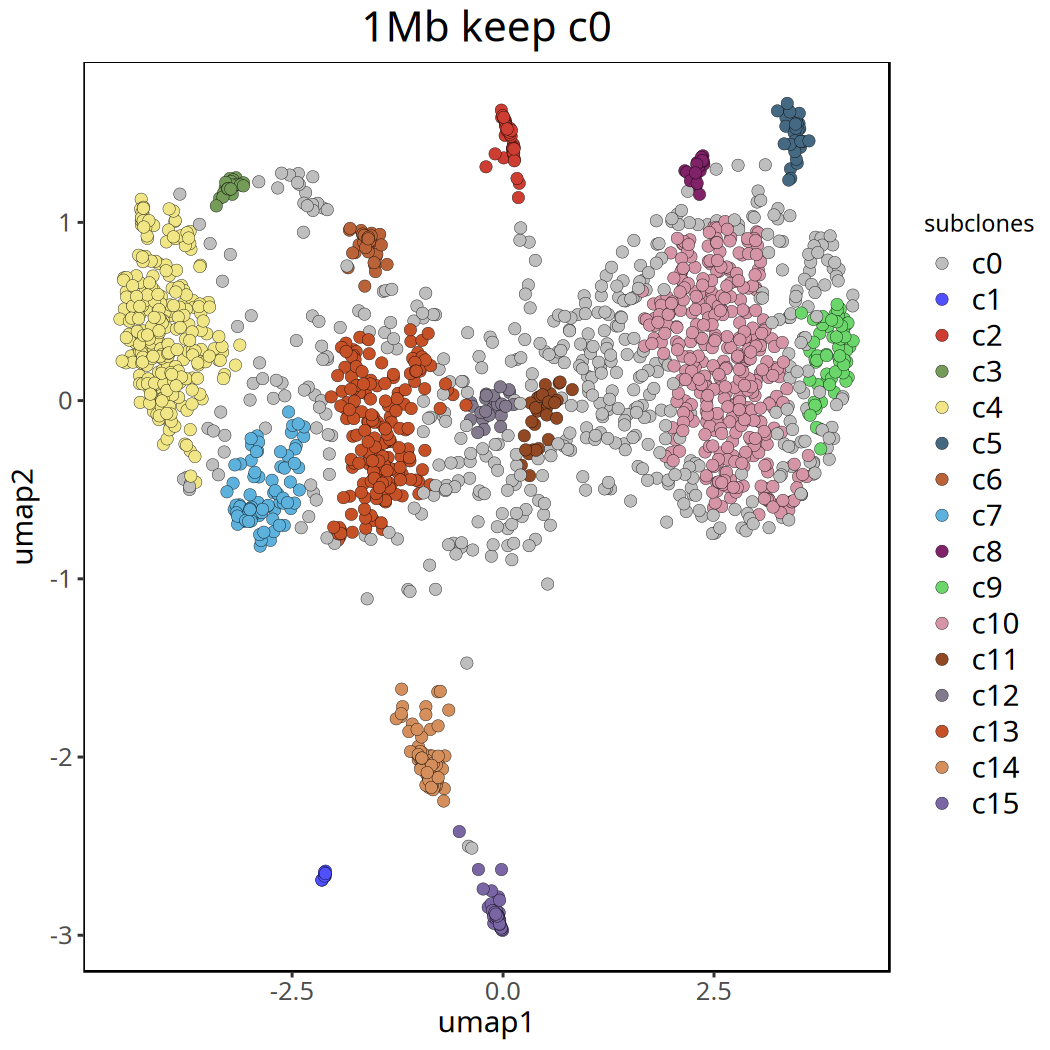
1Mb 分辨率下肿瘤亚克隆分析结果
以下展示了去除 c0 群前后的 UMAP 聚类图。
options(repr.plot.width = 7, repr.plot.height = 7)
pic <- plotUmap(tumor_list[['1Mb']], label = 'subclones') +
ggtitle('1Mb keep c0') +
theme(plot.title = element_text(hjust = .5, size = 20))
pic
ggsave(file.path(outdir, paste0('plotUmap_',samplename,'1Mb','_keep_c0.pdf')), pic, width = 7, height = 5)Coloring by: subclones.
options(repr.plot.width = 7, repr.plot.height = 7)
tumor_list[['1Mb']] <- tumor_list[['1Mb']][,colData(tumor_list[['1Mb']])$subclones != 'c0']
pic <- plotUmap(tumor_list[['1Mb']], label = 'subclones') +
ggtitle('1Mb') +
theme(plot.title = element_text(hjust = .5, size = 20))
pic
ggsave(file.path(outdir, paste0('plotUmap_',samplename,'_1Mb','_remove_c0.pdf')), pic, width = 7, height = 5)Coloring by: subclones.
1Mb 分辨率下肿瘤亚克隆 CNV 热图以及亚克隆进化树
options(repr.plot.width = 12, repr.plot.height = 6, repr.plot.res = 150)
tumor_list[['1Mb']] <- calcConsensus(tumor_list[['1Mb']])
tumor_list[['1Mb']] <- runConsensusPhylo(tumor_list[['1Mb']])
suppressMessages({
ht <- plotHeatmap(
tumor_list[['1Mb']],
label = 'subclones',
order_cells = 'consensus_tree'
)
})
ht
pdf(
file.path(outdir, paste0('plotHeatmap_consensus_tree_',samplename,'_1Mb','.pdf')),
width = 16,
height = 6)
ht
dev.off()pdf: 2
suppressWarnings({
pic <- plotPhylo(tumor_list[['1Mb']], label = 'subclones', consensus = TRUE)
})
pic
ggsave(file.path(outdir, paste0('plotPhylo_consensus_',samplename,'_1Mb','.pdf')), pic, width = 7, height = 7)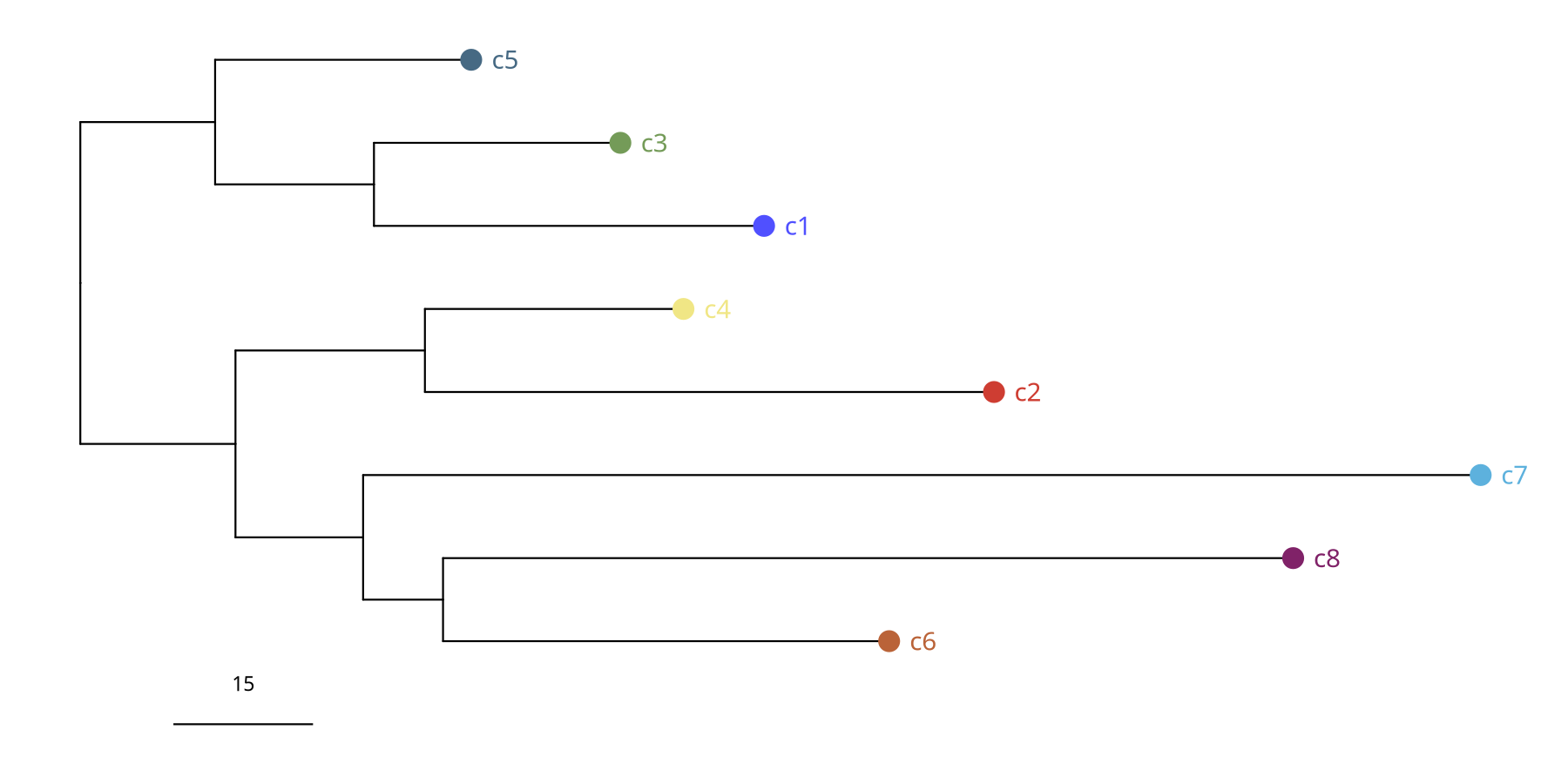
以下热图和折线图是从“Bulk”水平上观察不同亚克隆之间 CNV 的差异。
suppressMessages({
ht <- plotHeatmap(tumor_list[['1Mb']],consensus = TRUE,label = 'subclones')
})
ht
pdf(
file.path(outdir, paste0('plotHeatmap_consensus_',samplename,'_550kb','.pdf')),
width = 16,
height = 6)
ht
dev.off()pdf: 2
pic <- plotConsensusLine_static(tumor_list[['1Mb']],levels(colData(tumor_list[['1Mb']])$subclones) ) +
scale_color_manual(values = subclones_pal())
pic
ggsave(file.path(outdir, paste0('plot_ConsensusLine_',samplename,'_1Mb','.pdf')), pic, width = 7, height = 5)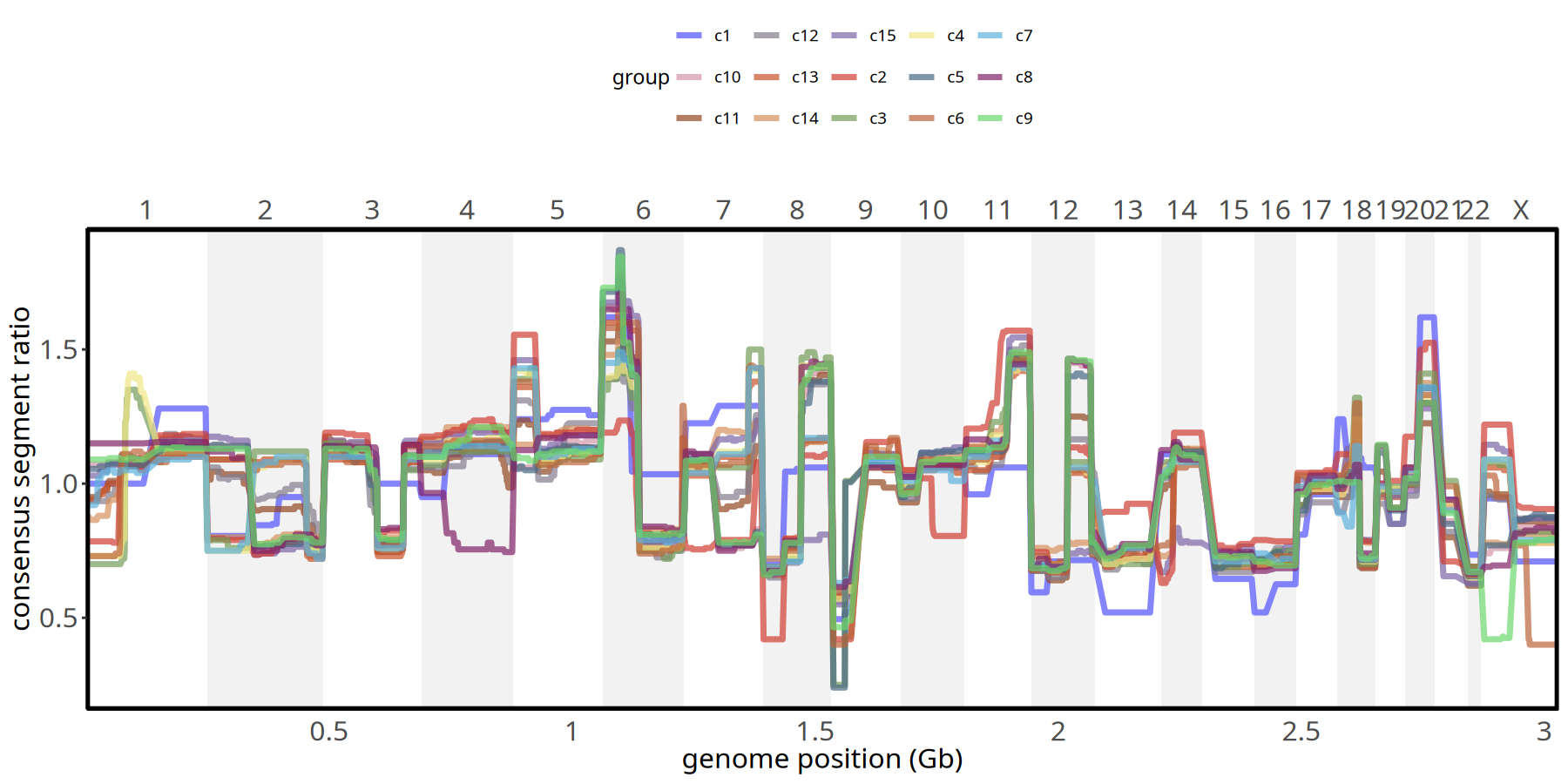
单细胞水平进化树
除了亚克隆层面的进化树,CopyKit 还可以通过 runPhylo 构建单细胞分辨率的进化树。这有助于观察克隆内部的微进化 (Micro-evolution) 过程。
- 原理:基于拷贝数图谱的相似性,计算细胞间的距离。拷贝数越相似,亲缘关系越近。
- 注意:由于单细胞数据量巨大且噪声较多,计算量较大。
220kb 分辨率单细胞进化树
图注:单细胞分辨率进化树。
- 末端节点:图中的每一个点代表一个独立的细胞。
- 颜色:标识该细胞所属的亚克隆。
- 意义:相比于亚克隆树,此图展示了亚克隆内部的结构,可用于观察细胞间的细微异质性。
suppressMessages({
tumor_list[['220kb']] <- runPhylo(tumor_list[['220kb']], metric = 'manhattan')
pic <- plotPhylo(tumor_list[['220kb']], label = 'subclones')
})pic
ggsave(file.path(outdir, paste0('plotPhylo_cells_',samplename,'_220kb.pdf')), width = 8, height = 6)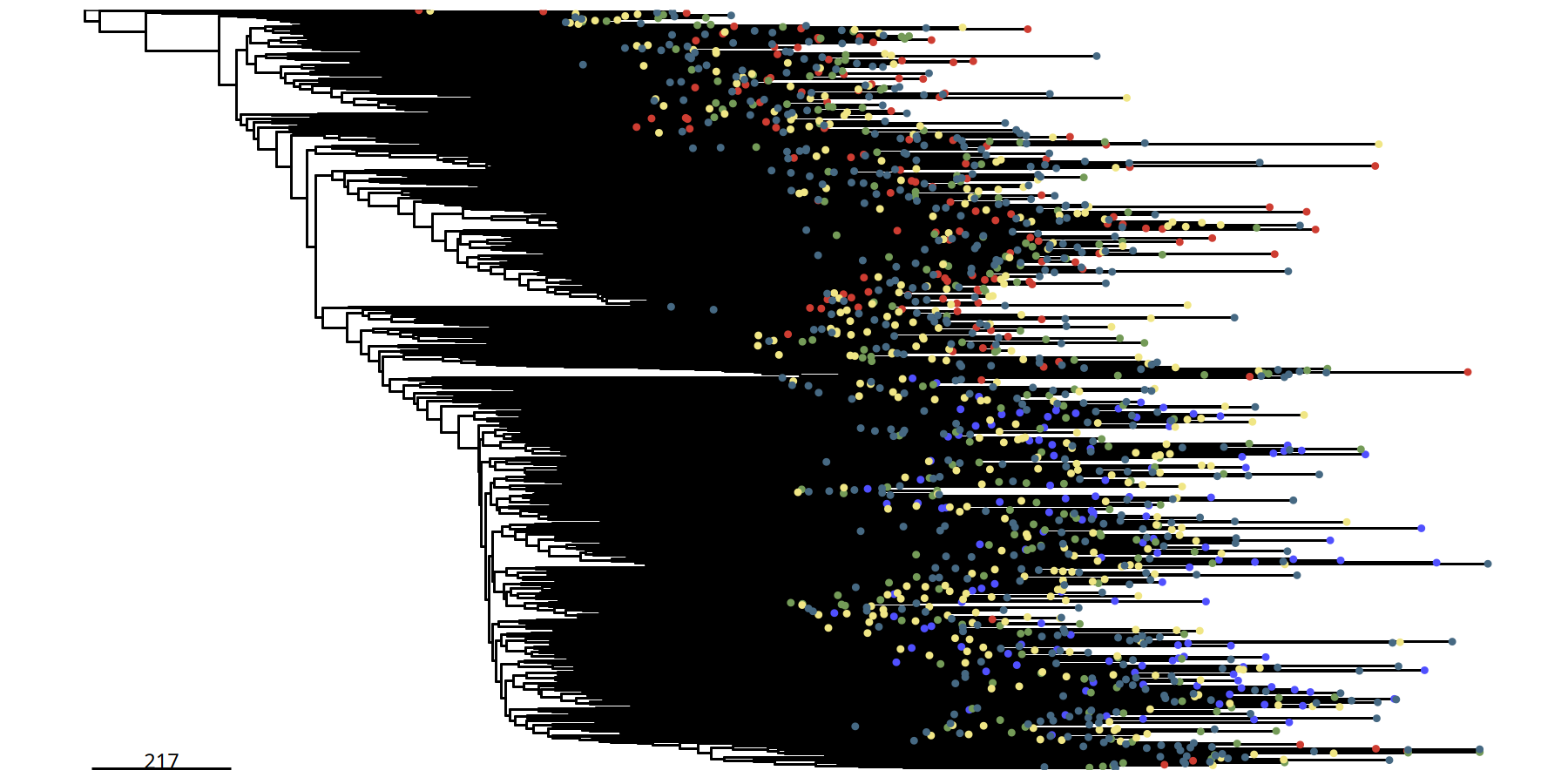
500kb 分辨率单细胞进化树
suppressMessages({
tumor_list[['500kb']] <- runPhylo(tumor_list[['500kb']], metric = 'manhattan')
pic <- plotPhylo(tumor_list[['500kb']], label = 'subclones')
})pic
ggsave(file.path(outdir, paste0('plotPhylo_cells_',samplename,'_500kb.pdf')), width = 8, height = 6)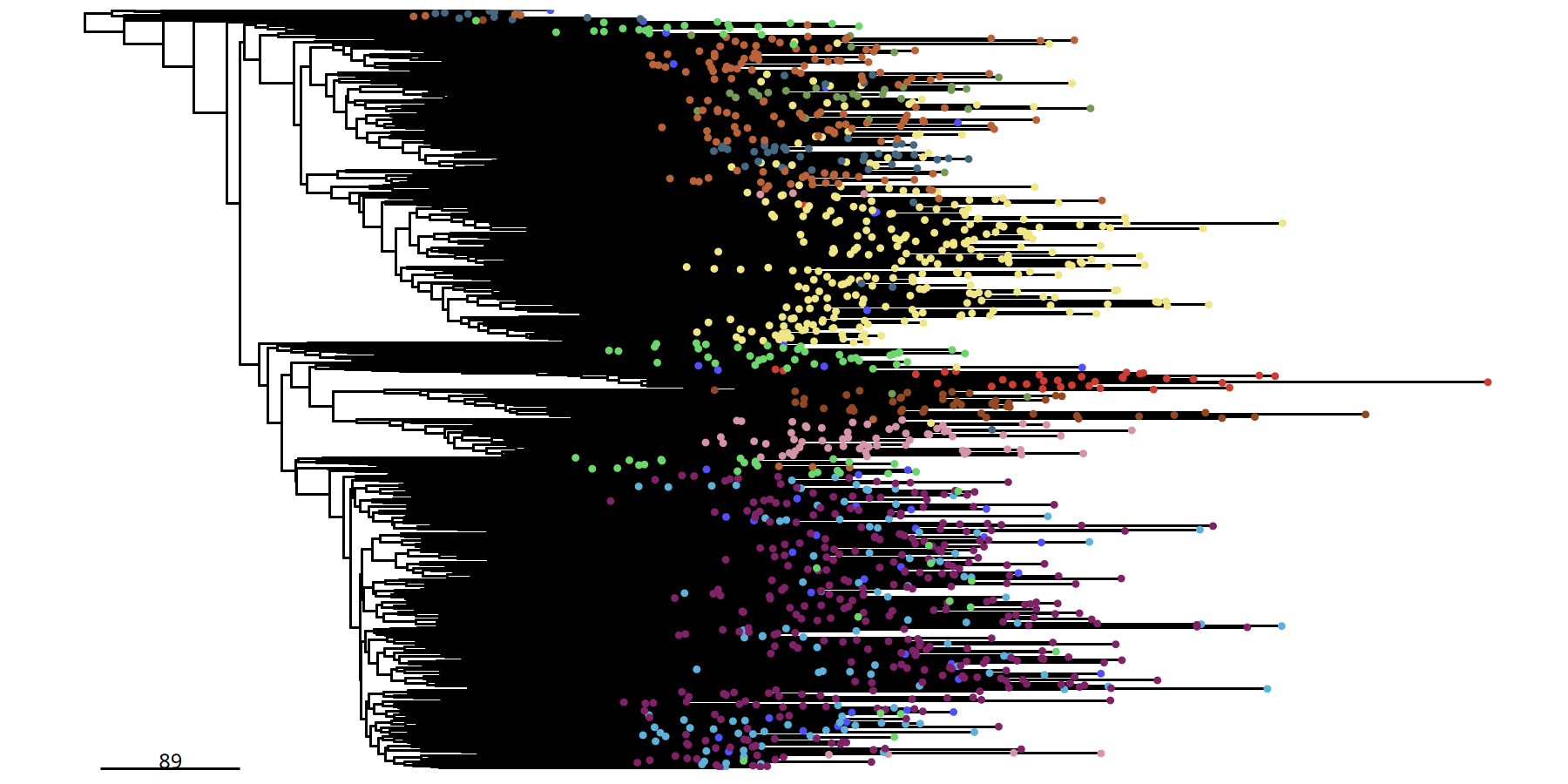
1Mb 分辨率单细胞进化树
suppressMessages({
tumor_list[['1Mb']] <- runPhylo(tumor_list[['1Mb']], metric = 'manhattan')
pic <- plotPhylo(tumor_list[['1Mb']], label = 'subclones')
})pic
ggsave(file.path(outdir, paste0('plotPhylo_cells_',samplename,'_1Mb.pdf')), width = 8, height = 6)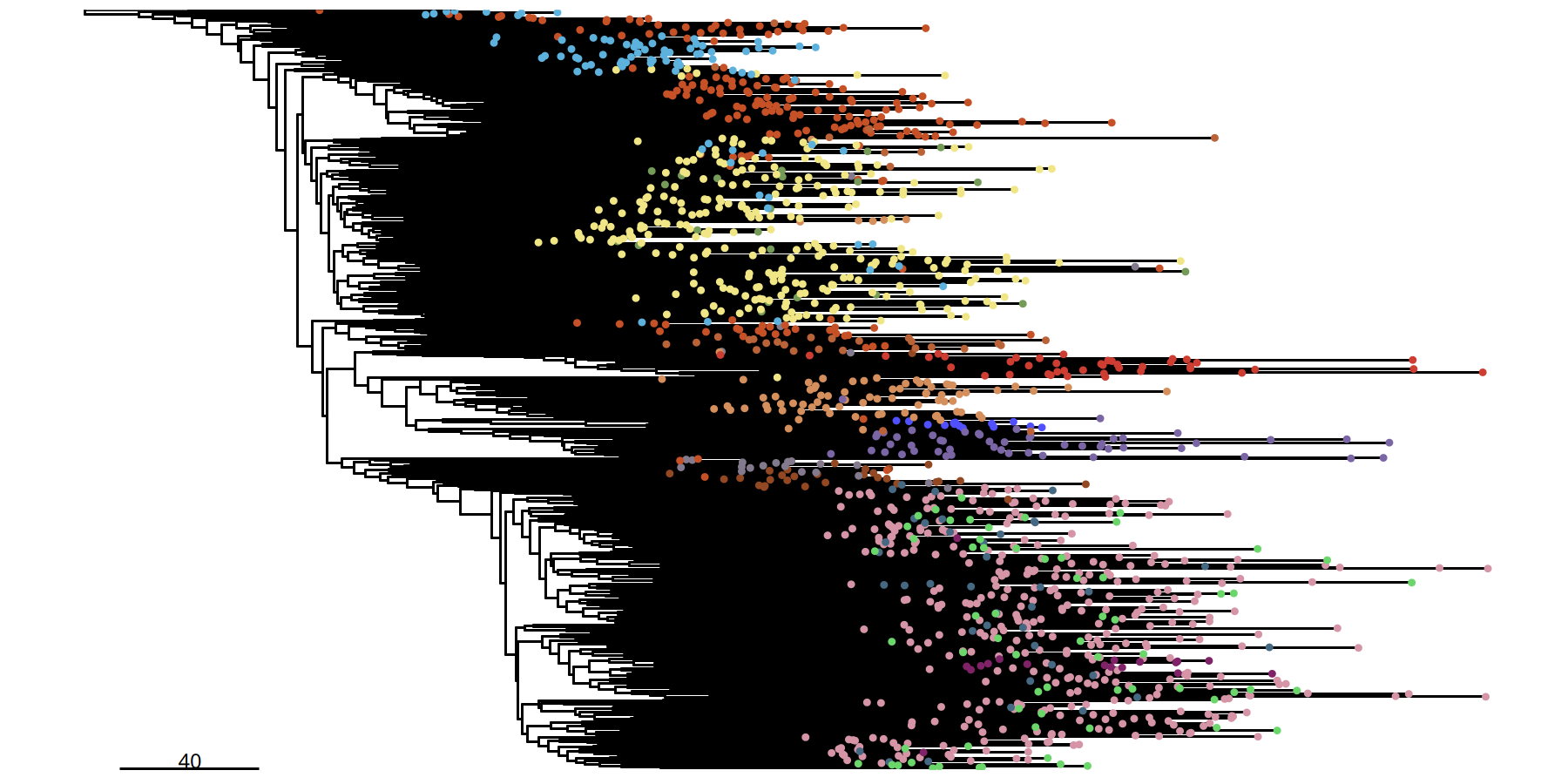
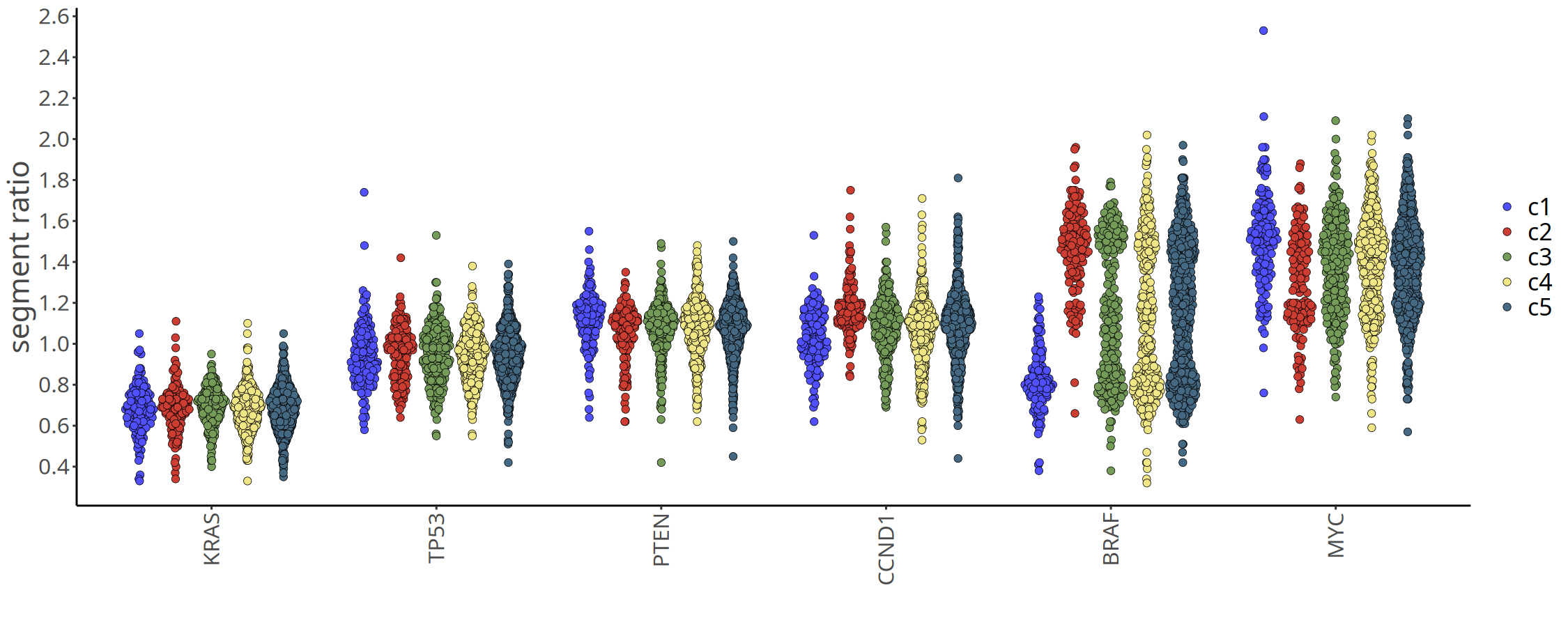
基因水平 CNV 可视化
除了全基因组图谱,我们通常关注特定驱动基因(如 MYC, TP53, EGFR 等)的拷贝数状态。plotGeneCopy 函数可以展示特定基因在不同亚克隆中的拷贝数分布。下图仅展示 220kb 分辨率下鉴定出的亚克隆的驱动基因拷贝数点图。
提示:您可以将列表中的基因替换为您感兴趣的靶点基因。 注意:如果某些基因位于 VarBin 算法的排除区域(如着丝粒、端粒或高重复区域),则无法被检测到(会显示警告信息)。
图注:驱动基因拷贝数点图 (Jitter Plot)。
- X 轴:不同的肿瘤亚克隆。
- Y 轴:推断的绝对整数拷贝数 (Integer Copy Number)。
- 含义:每个点代表一个细胞。该图直观展示了关键癌基因(如 MYC, EGFR)或抑癌基因(如 TP53, PTEN)在不同亚克隆中的扩增或缺失状态,有助于识别驱动肿瘤演化的关键变异事件。
options(repr.plot.height = 6, repr.plot.width = 15)
suppressMessages({
plotGeneCopy(
tumor_list[['220kb']],
genes = c("TP53","PTEN","MYC","CCND1","KRAS","BRAF"),
label = 'subclones',
dodge.width = 0.8)
})
saveRDS(tumor_list, file.path(outdir, paste0(samplename, '_copykit_analysis.rds')))更多功能:CopyKit 的更多高级可视化功能请详见 CopyKit 官方文档。