甲基化 + RNA 双组学: 双细胞判定流程
模块简介
本模块基于 ALLCools 和 MethylScrublet 算法开发,旨在从单细胞甲基化测序数据中识别并过滤双细胞 (doublets)。
双细胞是指在单细胞测序过程中,两个或多个细胞意外粘连并被作为一个"细胞"进行测序的技术伪影。双细胞会引入混合的表达/甲基化特征,严重干扰后续的细胞分群和差异分析(例如,可能错误地将双细胞鉴定为新的中间态细胞类型)。
本模块提供了三种互补的双细胞鉴定方法,可综合判断并提高识别准确性:
- 方法一:转录组细胞类型鉴定:基于 RNA 测序数据,通过检测不同细胞类型标志基因的异常共表达来识别双细胞。
- 方法二:甲基化 Reads 数鉴定:通过分析每个细胞的甲基化测序深度(Reads 数)分布,识别异常高覆盖度的潜在双细胞。
- 方法三:甲基化率鉴定 (MethylScrublet):使用 MethylScrublet 算法,通过模拟人造双细胞并与观测数据进行比对,计算每个细胞的"双细胞评分"。
TIP
本分析适用于单细胞多组学数据(同时包含 RNA 和甲基化信息),也支持仅使用甲基化数据进行双细胞识别。
输入文件准备
本模块需要以下输入文件:
必需文件
- 单细胞 RNA 测序数据:基因表达矩阵(
filtered_feature_bc_matrix目录)。 - 单细胞甲基化数据:MCDS 格式的甲基化数据集(
.mcds文件)。 - Barcode 映射文件(可选):如果试剂类型为 DD-MET3,用于关联 RNA 和甲基化数据的细胞 Barcode 对应关系。
文件结构示例
data/
├── AY1768874914782
│ └── methylation
│ └── demoWTJW969-task-1
│ └── WTJW969
│ ├── allcools_generate_datasets
│ │ └── WTJW969.mcds # 甲基化数据
│ ├── filtered_feature_bc_matrix # 转录组表达矩阵
│ └── split_bams
│ ├── WTJW969_cells.csv
│ └── filtered_barcode_reads_counts.csv
└── AY1768876253533
└── methylation
└── demo4WTJW880-task-1
└── WTJW880
├── allcools_generate_datasets
│ └── WTJW880.mcds # 甲基化数据
├── filtered_feature_bc_matrix # 转录组表达矩阵
└── split_bams
├── WTJW880_cells.csv
└── filtered_barcode_reads_counts.csv
DD-M_bUCB3_whitelist.csv # Barcode 映射文件(可选)# --- 导入必要的 Python 包 ---
# 系统操作和文件处理
import os
import re
import glob
import warnings
# ALLCools 相关包:用于甲基化数据处理和双细胞识别
from ALLCools.mcds import MCDS
from ALLCools.clustering import (
tsne, significant_pc_test, log_scale, lsi,
binarize_matrix, filter_regions, cluster_enriched_features,
ConsensusClustering, Dendrogram, get_pc_centers
)
from ALLCools.clustering.doublets import MethylScrublet
from ALLCools.plot import *
# 单细胞分析工具
import scanpy as sc
# 数据处理和可视化
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
from matplotlib.lines import Line2D
import seaborn as sns
from matplotlib.patches import Rectangle
# 其他工具
from scipy import sparse# --- 文件路径配置 ---
# 根据新的文件层级结构配置路径
# 格式:{样本名: {'top_dir': 顶层目录, 'demo_dir': demo目录}}
sample_path_config = {
"WTJW969": {"top_dir": "AY1768874914782", "demo_dir": "demoWTJW969-task-1"},
"WTJW880": {"top_dir": "AY1768876253533", "demo_dir": "demo4WTJW880-task-1"}
}
# 数据根目录(默认为 "data")
data_root = "data"
# 当前分析的样本名(可根据实际情况修改)
current_sample = "WTJW969"
## reagent_type:试剂盒类型('DD-MET3' 或 'DD-MET5')
## DD-MET3:RNA 和甲基化数据使用不同的 Barcode,需要通过映射文件关联
## DD-MET5:RNA 和甲基化数据使用相同的 Barcode,不需要映射文件
reagent_type = "DD-MET5"双细胞鉴定方法
本模块提供三种互补的双细胞鉴定方法,建议综合使用以提高识别准确性。
方法一:基于转录组细胞类型鉴定的双细胞识别
原理说明:
在单细胞 RNA 测序数据分析中,双细胞识别的一种常用策略是检测不同细胞类型标志基因的异常共表达现象。
判断逻辑:
当两个理论上应分别在不同细胞类型中表达的标志基因在同一细胞群体中同时呈现高表达时,提示该群体可能并非来源于单一细胞类型,而是由两个不同细胞类型的细胞共同构成的双细胞(即两个细胞在建库或测序过程中被误识为一个细胞)。
举例说明:
例如,当 T 细胞标志基因(如 CD3D)与 B 细胞标志基因(如 CD79A)在同一细胞群体中同时高表达时,该群体很可能代表由 T 细胞和 B 细胞组成的双细胞。
RNA 转录组数据预处理与降维聚类
warnings.filterwarnings('ignore')
# --- RNA 转录组数据预处理与降维聚类 ---
# 根据新的文件层级结构构建路径:data/{top_dir}/methylation/{demo_dir}/{sample}/filtered_feature_bc_matrix
top_dir = sample_path_config[current_sample]['top_dir']
demo_dir = sample_path_config[current_sample]['demo_dir']
rna_path = os.path.join('../../',data_root, top_dir, 'methylation', demo_dir, current_sample, 'filtered_feature_bc_matrix')
adata_rna = sc.read_10x_mtx(rna_path)
# 计算线粒体基因标记(用于质量控制)
adata_rna.var["mt"] = adata_rna.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(adata_rna, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True)
# 数据标准化:将每个细胞的总计数标准化到 10,000
sc.pp.normalize_total(adata_rna, target_sum=1e4)
# Log 转换:log(x + 1) 变换,使数据更接近正态分布
sc.pp.log1p(adata_rna)
# 识别高变基因:选择表达变异最大的 2000 个基因用于后续分析
sc.pp.highly_variable_genes(adata_rna, n_bins=100, n_top_genes=2000)
# 保存原始数据到 adata.raw,然后仅保留高变基因
adata_rna.raw = adata_rna
adata_rna = adata_rna[:, adata_rna.var.highly_variable]
# 标准化:Z-score 标准化,限制最大值为 10
sc.pp.scale(adata_rna, max_value=10)
# 主成分分析(PCA)降维
sc.tl.pca(adata_rna)
# 构建 KNN 图(用于 UMAP 和聚类)
sc.pp.neighbors(adata_rna)
# UMAP 降维可视化
sc.tl.umap(adata_rna)
# Leiden 聚类(分辨率 0.8)
sc.tl.leiden(adata_rna, resolution=0.8)
# 可视化 UMAP 结果,按聚类着色
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(6, 6))
sc.pl.umap(adata_rna, color='leiden', s=35, palette='Set2', frameon=True)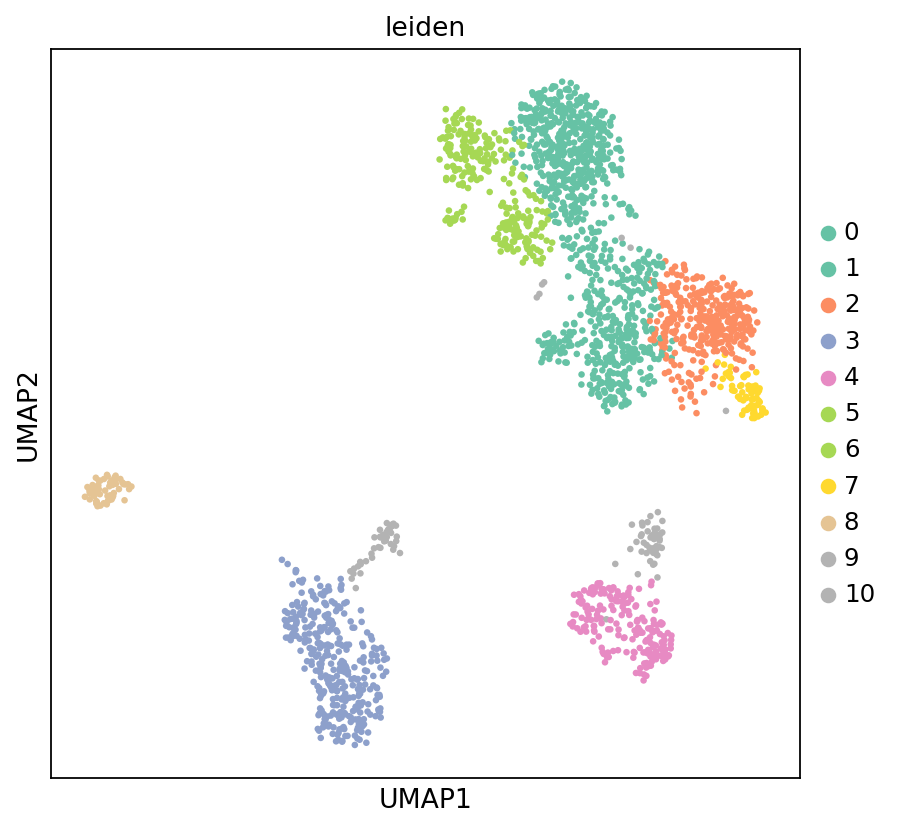
甲基化组数据预处理与降维聚类
# --- 甲基化组数据预处理与降维聚类 ---
# 根据新的文件层级结构构建路径:data/{top_dir}/methylation/{demo_dir}/{sample}/allcools_generate_datasets/{sample}.mcds
top_dir = sample_path_config[current_sample]['top_dir']
demo_dir = sample_path_config[current_sample]['demo_dir']
mcds_path = os.path.join('../../',data_root, top_dir, 'methylation', demo_dir, current_sample, 'allcools_generate_datasets', f'{current_sample}.mcds')
mcds = MCDS.open(
mcds_path,
obs_dim="cell",
var_dim="chrom20k"
)
# 提取甲基化评分矩阵(使用 hypo-score,即低甲基化评分)
adata_methy = mcds.get_score_adata(mc_type="CGN", quant_type="hypo-score")
# 二值化矩阵:将甲基化率 > 0.95 的区域标记为高甲基化
binarize_matrix(adata_methy, cutoff=0.95)
# 过滤低质量区域:移除变异度低或覆盖度不足的区域
filter_regions(adata_methy)
# LSI(Latent Semantic Indexing)降维:类似于 PCA,适用于稀疏矩阵
lsi(adata_methy, algorithm="arpack", obsm="X_lsi")
# 显著性主成分检验:仅保留 p < 0.1 的显著主成分
significant_pc_test(adata_methy, p_cutoff=0.1, obsm="X_lsi", update=True)
# 构建 KNN 图(使用 LSI 降维结果,邻居数 30)
sc.pp.neighbors(adata_methy, use_rep='X_lsi', n_neighbors=30)
# UMAP 降维可视化
sc.tl.umap(adata_methy)
# Leiden 聚类(分辨率 0.5)
sc.tl.leiden(adata_methy, resolution=0.5)126195 regions remained.
16 components passed P cutoff of 0.1.
Changing adata.obsm['X_pca'] from shape (2196, 100) to (2196, 16)
细胞类型标志基因表达分析
通过可视化不同细胞类型的标志基因表达模式,识别可能存在异常共表达的细胞群(潜在双细胞)。
# --- 可视化不同细胞类型的标志基因表达模式 ---
# 标志基因列表:
# - T 细胞:CD3D, CD8A, CD4
# - NK 细胞:NKG7
# - B 细胞:CD79A, MS4A1, JCHAIN
# - 单核/巨噬细胞:LYZ
# - 树突状细胞:LILRA4
dp = sc.pl.dotplot(
adata_rna,
["CD3D", "CD8A", "CD4", "NKG7", "CD79A", "MS4A1", "JCHAIN", "LYZ", "LILRA4"],
groupby='leiden',
standard_scale='var',
cmap='GnBu',
show=False
)
dp['mainplot_ax'].figure.set_size_inches(10, 6)
for label in dp['mainplot_ax'].get_xticklabels():
label.set_rotation(45)
label.set_ha('right')
label.set_rotation_mode('anchor')
dp['mainplot_ax'].set_title('Marker Genes Expression', pad=10, fontsize=12)
plt.show()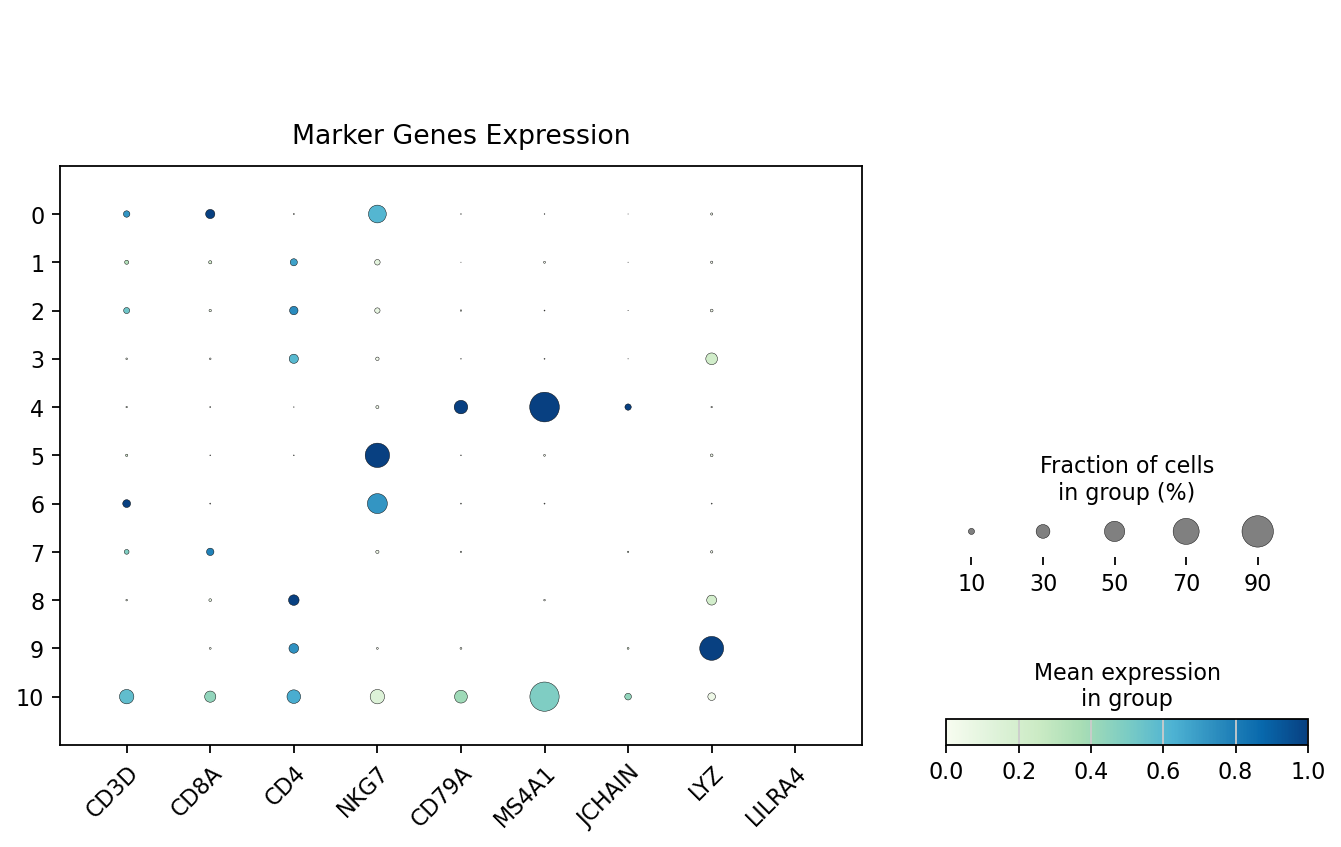
- 根据上述气泡图显示,
cluster 9可能为双细胞。该 cluster 同时表达多个不同细胞类型的标志基因(如 T 细胞标志基因和 B 细胞标志基因),呈现异常共表达模式,提示该 cluster 中的细胞可能是两个不同细胞类型的双细胞。
细胞 UMI 计数分布可视化
图注: 本图展示了所有细胞的 UMI 计数分布情况,用于识别异常高表达的潜在双细胞。
- 横坐标:按照 UMI 总数 (nCount_RNA) 从高到低排序的细胞顺序。
- 纵坐标:对应细胞的转录本总量(nCount_RNA,UMI 总数)的以 2 为底的对数转换值 (log2)。数值越高表示该细胞检测到的 RNA 丰度越高。
- 中间散点图:每个点代表一个细胞,不同颜色表示不同的细胞群(Leiden 聚类)。位于排序前端且 UMI 数显著偏高的细胞可能代表异常高表达细胞,提示潜在的双细胞或多细胞事件。
- 上方密度曲线:展示不同细胞群体整体的 nCount_RNA 分布特征,不同颜色对应不同的 Leiden 聚类,其峰值反映各细胞群体的典型 RNA 丰度水平。
- 右侧密度曲线:按细胞群体展示 log2(nCount_RNA) 的总体分布,用于比较不同细胞群体在 RNA 丰度上的差异。
warnings.filterwarnings('ignore')
MYCOLOR = None
plot_data = adata_rna.obs[["total_counts", "leiden"]]
plot_data = plot_data.sort_values('total_counts', ascending=False).reset_index(drop=True)
plot_data['order'] = plot_data.index + 1
plot_data['log2_nCount_RNA'] = np.log2(plot_data['total_counts'] + 1)
# 创建图形 - 缩小图片尺寸,便于在 Jupyter 中查看代码
fig = plt.figure(figsize=(10, 6), dpi=80)
# 使用GridSpec,将图例放在右侧,避免遮挡 x 轴标签
gs = fig.add_gridspec(2, 3,
width_ratios=[7, 2, 2.5], # 主图、右侧密度图、图例(增大图例区域)
height_ratios=[2, 6], # 顶部密度图、主图
hspace=0.1, wspace=0.1) # 增加上下间距,增加左右间距,避免图例挤着主图
ax_main = fig.add_subplot(gs[1, 0])
ax_top = fig.add_subplot(gs[0, 0], sharex=ax_main)
ax_right = fig.add_subplot(gs[1, 1], sharey=ax_main)
ax_legend = fig.add_subplot(gs[:, 2]) # 图例放在右侧整列
# 隐藏轴标签
ax_top.tick_params(axis='x', labelbottom=False)
ax_right.tick_params(axis='y', labelleft=False)
ax_top.grid(False)
ax_right.grid(False)
# 获取颜色调色板
palette = sns.color_palette('tab20', len(plot_data['leiden'].unique()))
color_map = dict(zip(plot_data['leiden'].unique(), palette))
# 1. 主散点图
scatter = sns.scatterplot(
data=plot_data,
x='order',
y='log2_nCount_RNA',
hue='leiden',
palette=MYCOLOR if MYCOLOR else color_map,
s=15,
alpha=0.5,
edgecolor=None,
ax=ax_main,
linewidth=0.5,
legend=False # 不在主图上显示图例
)
# 设置网格
ax_main.grid(True, linestyle='--', alpha=0.3)
# 设置X轴标签
ax_main.set_xlabel('Order(Sorted_by_Total_Counts)', fontsize=12)
ax_main.set_ylabel('log2(Total_Counts)', fontsize=12)
# 2. 顶部密度图(截断小于0的部分)
for celltype, color in color_map.items():
celltype_data = plot_data[plot_data['leiden'] == celltype]
# 计算密度
if len(celltype_data) > 1:
from scipy.stats import gaussian_kde
# 使用KDE计算密度
kde = gaussian_kde(celltype_data['order'])
x_density = np.linspace(celltype_data['order'].min(), celltype_data['order'].max(), 200)
y_density = kde(x_density)
# 将密度值标准化并截断负值
y_density = np.maximum(y_density, 0)
# 绘制填充(从0开始)
ax_top.fill_between(x_density, 0, y_density,
color=color, alpha=0.5)
# 绘制密度线
ax_top.plot(x_density, y_density,
color=color, alpha=0.7, linewidth=1)
# 设置顶部密度图的y轴下限为0
ax_top.set_ylim(bottom=0)
ax_top.set_ylabel('Density', fontsize=12)
# 3. 右侧密度图(截断小于0的部分)
for celltype, color in color_map.items():
celltype_data = plot_data[plot_data['leiden'] == celltype]
# 计算密度
if len(celltype_data) > 1:
from scipy.stats import gaussian_kde
# 使用KDE计算密度
kde = gaussian_kde(celltype_data['log2_nCount_RNA'])
y_density = np.linspace(celltype_data['log2_nCount_RNA'].min(),
celltype_data['log2_nCount_RNA'].max(), 200)
x_density = kde(y_density)
# 将密度值标准化并截断负值
x_density = np.maximum(x_density, 0)
# 绘制填充(从0开始)
ax_right.fill_betweenx(y_density, 0, x_density,
color=color, alpha=0.5)
# 绘制密度线
ax_right.plot(x_density, y_density,
color=color, alpha=0.7, linewidth=1)
# 设置右侧密度图的x轴下限为0
ax_right.set_xlim(left=0)
ax_right.set_xlabel('Density', fontsize=12)
# 4. 在底部创建图例
# 获取唯一的leiden值和对应的颜色
unique_labels = plot_data['leiden'].unique()
leiden_labels = sorted(unique_labels, key=lambda x: int(x))
colors = [color_map[label] for label in leiden_labels]
# 创建图例的handles
handles = [plt.Line2D([0], [0], marker='o', color='w',
markerfacecolor=color, markersize=4,
label=label, alpha=0.7)
for label, color in zip(leiden_labels, colors)]
# 计算图例的列数(根据leiden数量自动调整)
n_cols = min(8, len(leiden_labels)) # 最多8列
# 在右侧轴创建图例
ax_legend.axis('off') # 关闭右侧轴的坐标轴
# 将图例放在右侧,留出更多空间
legend = ax_legend.legend(handles=handles,
loc='center left', # 左侧居中
bbox_to_anchor=(0.1, 0.5), # 图例位置,留出左边距
ncol=1, # 单列显示
fontsize=12,
title='Cluster',
title_fontsize=12,
frameon=False,
fancybox=True,
framealpha=0.8,
alignment='left')
# 调整布局 - 图例在右侧,不需要额外底部边距
plt.tight_layout(rect=[0, 0.05, 1, 0.95]) # 正常边距
plt.show()
# 限制图片显示大小,避免占满整个视图
plt.rcParams['figure.max_open_warning'] = 0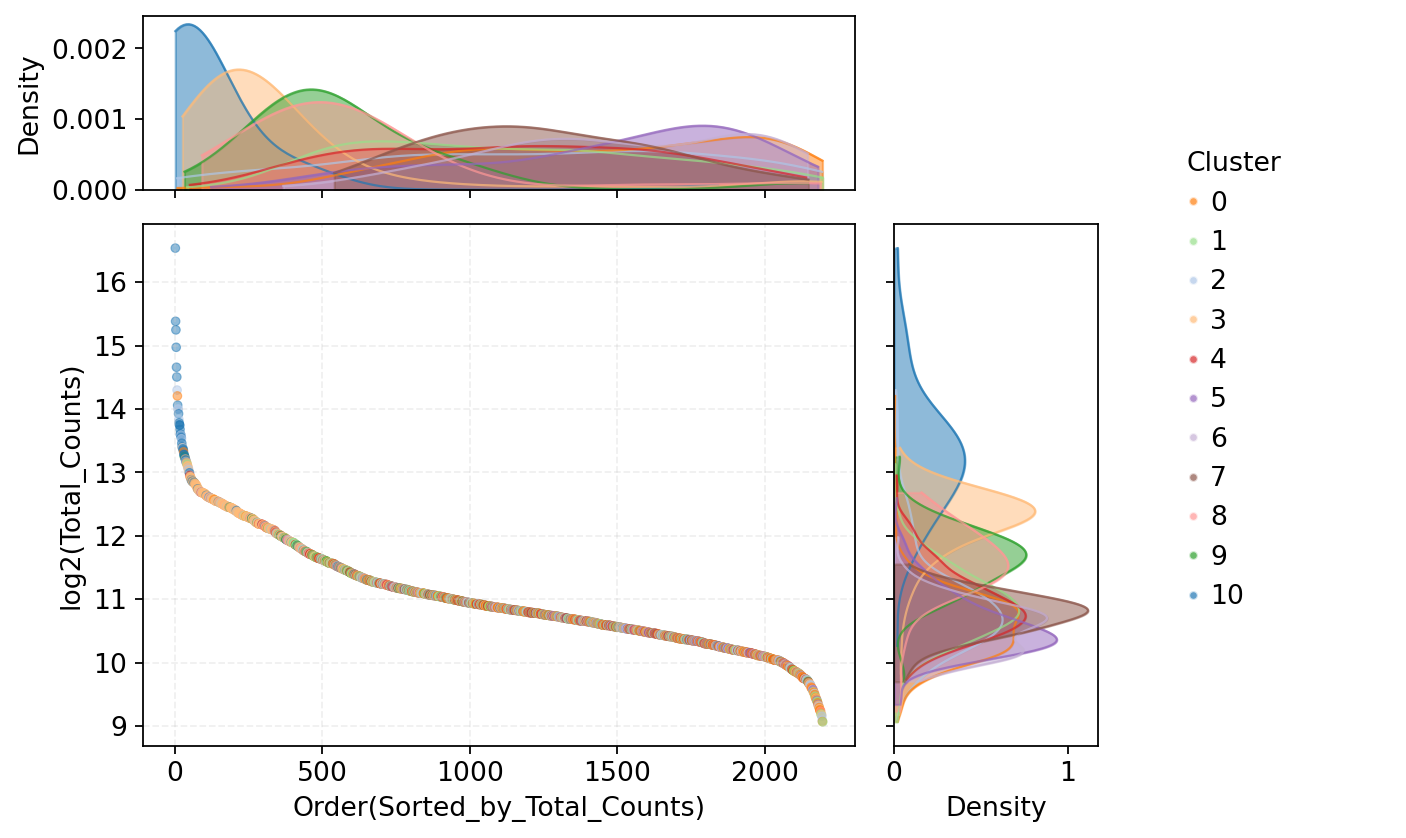
方法二:基于甲基化 Reads 数的双细胞识别
原理说明:
由于双细胞包含来自两个细胞的 DNA,其甲基化测序深度(Reads 数)通常显著高于单一细胞。通过分析单细胞甲基化数据中各细胞的 Reads 数分布,可用于识别测序覆盖度异常偏高的潜在双细胞。
判断逻辑:
- 双细胞的 Reads 数通常显著高于同类单细胞的平均水平,在理想情况下可接近单细胞 Reads 数的两倍。
- 在按 Reads 数排序的分布图中,双细胞往往集中于高 Reads 数区间。
- 通过设定合理的 Reads 数阈值,可将上述覆盖度异常偏高的细胞识别并过滤。
# --- 读取每个细胞的甲基化 Reads 计数文件 ---
# 根据新的文件层级结构构建路径:data/{top_dir}/methylation/{demo_dir}/{sample}/split_bams/filtered_barcode_reads_counts.csv
top_dir = sample_path_config[current_sample]['top_dir']
demo_dir = sample_path_config[current_sample]['demo_dir']
reads_count_path = os.path.join('../../',data_root, top_dir, 'methylation', demo_dir, current_sample, 'split_bams', 'filtered_barcode_reads_counts.csv')
read_count = pd.read_csv(reads_count_path, header=0)
# 提取 RNA 数据的细胞类型信息
rna_meta = pd.DataFrame(adata_rna.obs["leiden"])
rna_meta["gex_cb"] = rna_meta.indexTIP
根据您使用的试剂盒类型,请选择相应的运行流程:
DD-MET5 试剂盒:
- 跳过下面的单元格(DD-MET3 专用)
- 直接运行后续单元格中的
if reagent_type == "DD-MET3"...代码
DD-MET3 试剂盒:
- 必须先运行下面的单元格(读取 Barcode 映射文件并合并)
- 然后再运行后续的
if reagent_type == "DD-MET3"...代码
# --- 读取 RNA 和甲基化数据的 Barcode 映射文件 ---
# 请先将映射文件下载到本地 https://github.com/seekgene/SeekSoulMethyl/blob/nf_rna_methy/nf/bin/barcodes/DD-M_bUCB3_whitelist.csv
gex_mc_bc_map = pd.read_csv(
"DD-M_bUCB3_whitelist.csv",
index_col=None
)
# 统一 Barcode 列名
gex_mc_bc_map = gex_mc_bc_map.rename(columns={'m_cb': 'barcode'})
# 合并数据:将 Reads 计数与 Barcode 映射和细胞类型信息关联
read_count = read_count.merge(gex_mc_bc_map)if reagent_type == "DD-MET3":
read_count = read_count.merge(rna_meta)
elif reagent_type == "DD-MET5":
rna_meta = rna_meta.rename(columns = {'gex_cb': 'barcode'})
read_count = read_count.merge(rna_meta)每个细胞的甲基化 Reads 数分布可视化
图注: 本图展示了所有细胞的甲基化 Reads 数分布,用于识别测序覆盖度异常偏高的潜在双细胞
- 横坐标:按照 Reads 总数从高到低排序的细胞顺序。
- 纵坐标:对应细胞的 Reads 总数经以 2 为底的对数转换值 (log2)。数值越高表示该细胞的甲基化测序覆盖度越高。
- 中间散点图:每个点代表一个细胞,不同颜色表示不同的细胞类型。位于排序前端且 Reads 数显著偏高的细胞提示存在异常高覆盖度,可能对应双细胞或多细胞事件。
- 上方密度曲线:展示不同细胞类型整体的 Reads 数分布特征,不同颜色对应不同细胞类型,其峰值反映各细胞类型的典型测序覆盖水平。
- 右侧密度曲线:按细胞类型展示 log2(Reads 数) 的总体分布,用于比较不同细胞类型在测序覆盖度上的差异。
warnings.filterwarnings('ignore')
MYCOLOR = None
read_count = read_count.sort_values('reads_counts', ascending=False).reset_index(drop=True)
read_count['order'] = read_count.index + 1
read_count['log2_reads_counts'] = np.log2(read_count['reads_counts'] + 1)
# 创建图形 - 缩小图片尺寸,便于在 Jupyter 中查看代码
fig = plt.figure(figsize=(10, 6), dpi=80) # 缩小图片尺寸,不占满整个视图
# 使用GridSpec,将图例放在右侧,避免遮挡 x 轴标签
gs = fig.add_gridspec(2, 3,
width_ratios=[7, 2, 2.5], # 主图、右侧密度图、图例(增大图例区域)
height_ratios=[2, 6], # 顶部密度图、主图
hspace=0.1, wspace=0.1) # 增加上下间距,增加左右间距,避免图例挤着主图
ax_main = fig.add_subplot(gs[1, 0])
ax_top = fig.add_subplot(gs[0, 0], sharex=ax_main)
ax_right = fig.add_subplot(gs[1, 1], sharey=ax_main)
ax_legend = fig.add_subplot(gs[:, 2]) # 图例放在右侧整列
ax_top.grid(False)
ax_right.grid(False)
# 隐藏轴标签
ax_top.tick_params(axis='x', labelbottom=False)
ax_right.tick_params(axis='y', labelleft=False)
# 获取颜色调色板
palette = sns.color_palette('tab20', len(read_count['leiden'].unique()))
color_map = dict(zip(read_count['leiden'].unique(), palette))
# 1. 主散点图
scatter = sns.scatterplot(
data=read_count,
x='order',
y='log2_reads_counts',
hue='leiden',
palette=MYCOLOR if MYCOLOR else color_map,
s=8,
alpha=0.7,
ax=ax_main,
edgecolor=None,
linewidth=0.5,
legend=False # 不在主图上显示图例
)
# 设置网格
ax_main.grid(True, linestyle='--', alpha=0.3)
# 设置X轴标签
ax_main.set_xlabel('Order(Sorted_by_Reads_Counts)', fontsize=12)
ax_main.set_ylabel('log2(Reads_Counts)', fontsize=12)
# 2. 顶部密度图(截断小于0的部分)
for celltype, color in color_map.items():
celltype_data = read_count[read_count['leiden'] == celltype]
# 计算密度
if len(celltype_data) > 1:
from scipy.stats import gaussian_kde
# 使用KDE计算密度
kde = gaussian_kde(celltype_data['order'])
x_density = np.linspace(celltype_data['order'].min(), celltype_data['order'].max(), 200)
y_density = kde(x_density)
# 将密度值标准化并截断负值
y_density = np.maximum(y_density, 0)
# 绘制填充(从0开始)
ax_top.fill_between(x_density, 0, y_density,
color=color, alpha=0.5)
# 绘制密度线
ax_top.plot(x_density, y_density,
color=color, alpha=0.7, linewidth=1)
# 设置顶部密度图的y轴下限为0
ax_top.set_ylim(bottom=0)
ax_top.set_ylabel('Density', fontsize=12)
# 3. 右侧密度图(截断小于0的部分)
for celltype, color in color_map.items():
celltype_data = read_count[read_count['leiden'] == celltype]
# 计算密度
if len(celltype_data) > 1:
from scipy.stats import gaussian_kde
# 使用KDE计算密度
kde = gaussian_kde(celltype_data['log2_reads_counts'])
y_density = np.linspace(celltype_data['log2_reads_counts'].min(),
celltype_data['log2_reads_counts'].max(), 200)
x_density = kde(y_density)
# 将密度值标准化并截断负值
x_density = np.maximum(x_density, 0)
# 绘制填充(从0开始)
ax_right.fill_betweenx(y_density, 0, x_density,
color=color, alpha=0.5)
# 绘制密度线
ax_right.plot(x_density, y_density,
color=color, alpha=0.7, linewidth=1)
# 设置右侧密度图的x轴下限为0
ax_right.set_xlim(left=0)
ax_right.set_xlabel('Density', fontsize=12)
# 4. 在底部创建图例
# 获取唯一的leiden值和对应的颜色
unique_labels = plot_data['leiden'].unique()
leiden_labels = sorted(unique_labels, key=lambda x: int(x))
colors = [color_map[label] for label in leiden_labels]
# 创建图例的handles
handles = [plt.Line2D([0], [0], marker='o', color='w',
markerfacecolor=color, markersize=4,
label=label, alpha=0.7)
for label, color in zip(leiden_labels, colors)]
# 计算图例的列数(根据leiden数量自动调整)
n_cols = min(8, len(leiden_labels)) # 最多8列
# 在右侧轴创建图例
ax_legend.axis('off') # 关闭右侧轴的坐标轴
# 将图例放在右侧,留出更多空间
legend = ax_legend.legend(handles=handles,
loc='center left', # 左侧居中
bbox_to_anchor=(0.1, 0.5), # 图例位置,留出左边距
ncol=1, # 单列显示
fontsize=12,
title='Clusters',
title_fontsize=12,
frameon=False,
fancybox=True,
framealpha=0.8,
alignment='left')
# 调整布局 - 图例在右侧,不需要额外底部边距
plt.tight_layout(rect=[0, 0.05, 1, 0.95]) # 正常边距
plt.show()
# 限制图片显示大小,避免占满整个视图
plt.rcParams['figure.max_open_warning'] = 0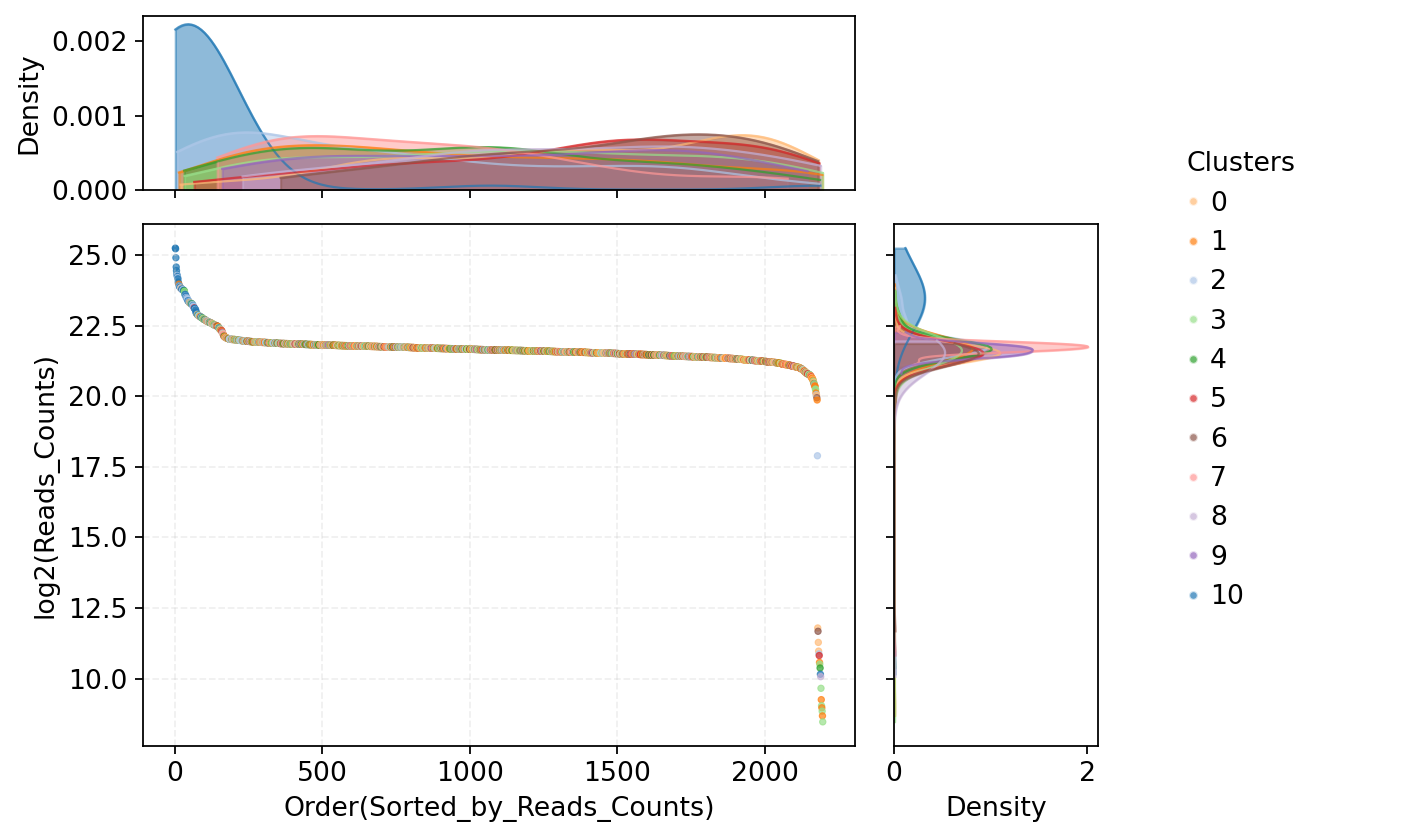
细胞 Reads 数相对变化分析
图注: 本图展示了细胞 Reads 数的相对变化分析,用于识别 Reads 数分布中的拐点,从而确定双细胞及低质量细胞的筛选阈值。
- 横轴:按照 Reads 总数从高到低排序的细胞顺序。左侧为 Reads 数异常偏高的细胞,右侧为 Reads 数偏低的细胞,中间区域对应中等测序覆盖度的细胞。
- 纵轴:相邻细胞在 Reads 数排序中的相对变化幅度,定义为
(pre - post) / pre。数值接近 0 表示相邻细胞 Reads 数变化较小,数值越大表示变化越显著。- 红色虚线:表示选定的 Reads 数阈值范围。左侧阈值用于排除 Reads 数异常偏高的细胞(潜在双细胞),右侧阈值用于排除 Reads 数过低的低质量细胞。两条虚线之间的细胞 Reads 数变化相对平稳,适用于后续分析。
# --- 准备数据:按 Reads 数降序排序 ---
p4_data = read_count.copy()
# 按 reads_counts 降序排序
p4_data = p4_data.sort_values('reads_counts', ascending=False).reset_index(drop=True)
p4_data['order'] = p4_data.index + 1
# --- 计算相邻细胞的相对变化:value = (pre - post) / pre ---
plot_df = pd.DataFrame({
'order': range(1, len(p4_data)),
'value': (p4_data['reads_counts'].iloc[:-1].values - p4_data['reads_counts'].iloc[1:].values) /
p4_data['reads_counts'].iloc[:-1].values
})# --- 可视化 Reads 数相对变化分布(带 LOESS 平滑) ---
plt.figure(figsize=(8, 6), dpi=70)
# 原始数据线图
plt.plot(plot_df['order'], plot_df['value'], alpha=0.7, linewidth=1, label='Raw data')
# LOESS 平滑(使用移动平均模拟)
span = 0.1
window_size = max(1, int(len(plot_df) * span))
smoothed = plot_df['value'].rolling(window=window_size, center=True, min_periods=1).mean()
# 绘制平滑后的曲线
plt.plot(plot_df['order'], smoothed, color='red', linewidth=1.5, label='LOESS Smoothed')
# 添加阈值线(可根据实际情况调整)
plt.axvline(x=200, linestyle='--', color='red', alpha=0.7, label='High Reads Threshold')
plt.axvline(x=2000, linestyle='--', color='red', alpha=0.7, label='Low Reads Threshold')
plt.xlabel('Order(Sorted_by_Reads_Count)', fontsize=12)
plt.ylabel('Relative_Difference\n(pre-post)/pre', fontsize=12)
plt.title('Reads Distribution with LOESS Smoothing', fontsize=12)
plt.legend(fontsize=12)
plt.grid(True, alpha=0.3)
plt.tight_layout()
plt.show()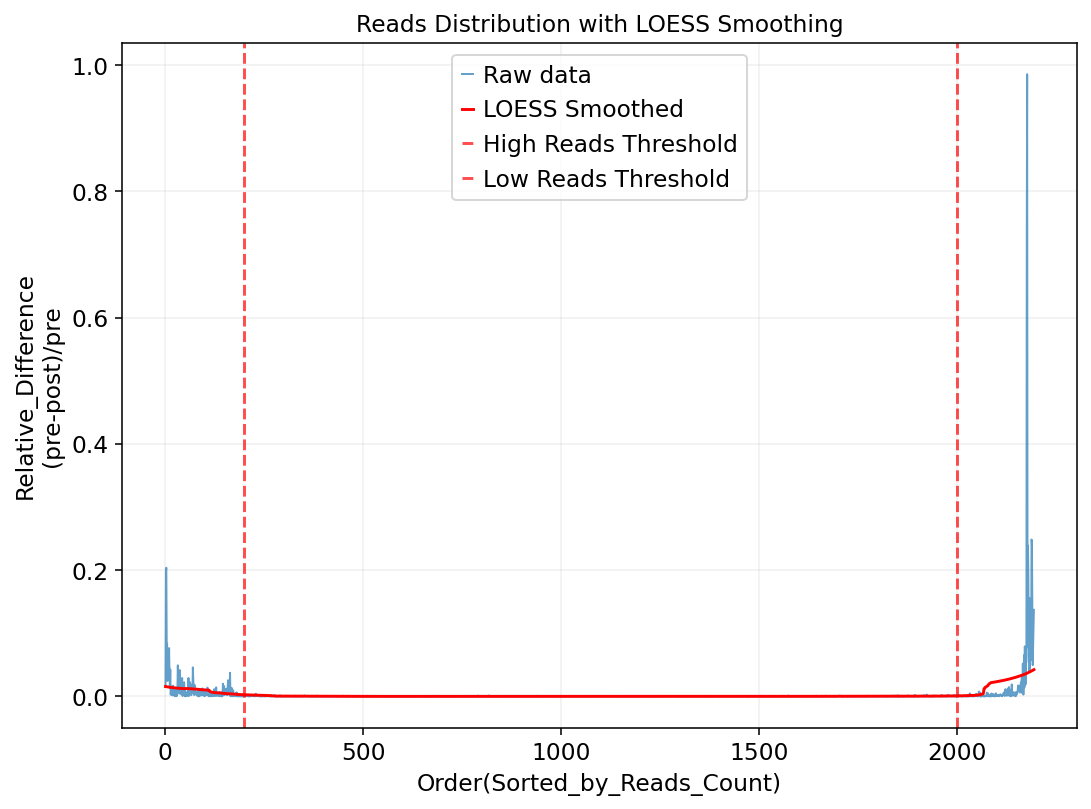
高/低 Reads 细胞 UMAP 高亮
图注: 本图在 UMAP 降维空间中高亮显示潜在的双细胞和空细胞,用于评估这些异常细胞在细胞图谱中的空间分布特征。
- 左图 (Potential Multi-Cells):在 UMAP 空间中高亮显示潜在双细胞。其中,红色点表示 Reads 数最高的前 200 个细胞(候选双细胞),浅灰色点表示其余正常细胞。通过观察红色点在 UMAP 空间中的分布情况,可评估潜在双细胞是否集中于特定区域,或分散分布于整体细胞图谱中。
- 右图 (Potential Empty Cells):在 UMAP 空间中高亮显示潜在空细胞。其中,红色点表示 Reads 数最低的后 200 个细胞(候选空细胞),浅灰色点表示其余正常细胞。该图用于评估潜在空细胞的空间分布模式,并判断其是否对应特定的低质量细胞群体。
warnings.filterwarnings('ignore')
# --- 根据 Reads 数阈值识别潜在双细胞和空细胞 ---
# 获取 Reads 数最高的 1000 个细胞(潜在双细胞)
multi_cells = p4_data.head(200)['barcode'].tolist()
# 获取 Reads 数最低的 1000 个细胞(潜在空细胞)
empty_cells = p4_data.tail(200)['barcode'].tolist()
# --- 在 AnnData 中创建标记列 ---
adata_methy.obs['cell_multi_highlight'] = 'other'
adata_methy.obs['cell_empty_highlight'] = 'other'
adata_methy.obs.loc[adata_methy.obs.index.isin(multi_cells), 'cell_multi_highlight'] = 'multi_cells'
adata_methy.obs.loc[adata_methy.obs.index.isin(empty_cells), 'cell_empty_highlight'] = 'empty_cells'
# --- 在 UMAP 空间中可视化潜在双细胞和空细胞的分布 ---
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(12, 5), dpi=80)
# 子图1: 高亮潜在双细胞(红色)
sc.pl.umap(
adata_methy,
color='cell_multi_highlight',
palette={'multi_cells': 'red', 'other': 'lightgrey'},
ax=ax1,
s=25,
show=False,
title='Potential Multi-Cells (High Read Counts)'
)
# 子图2: 高亮潜在空细胞(红色)
sc.pl.umap(
adata_methy,
color='cell_empty_highlight',
palette={'empty_cells': 'red', 'other': 'lightgrey'},
ax=ax2,
s=25,
show=False,
title='Potential Empty Cells (Low Read Counts)'
)
plt.tight_layout()
plt.show()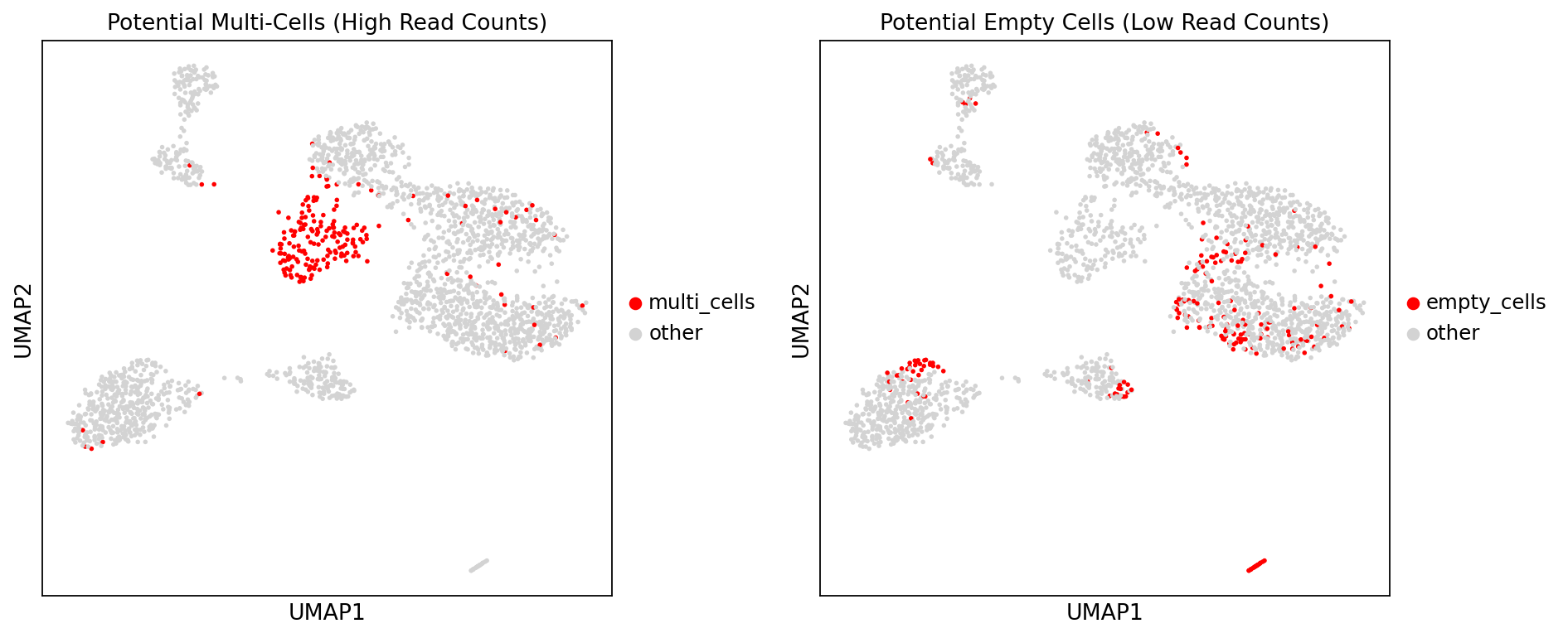
# --- 根据试剂类型将RNA注释映射到甲基化数据 ---
if reagent_type == "DD-MET3":
# DD-MET3: 通过map表将注释映射到adata_methyl
# gex_mc_bc_map包含:g_cb(RNA barcode)和barcode(甲基化barcode,原m_cb)
# 创建RNA注释的DataFrame
rna_annotation = pd.DataFrame({
'g_cb': adata_rna.obs.index,
'rna_leiden': adata_rna.obs['leiden'].values
})
# 通过映射表将RNA注释关联到甲基化barcode
# gex_mc_bc_map: g_cb -> barcode (甲基化barcode)
annotation_map = rna_annotation.merge(
gex_mc_bc_map[['g_cb', 'barcode']],
on='g_cb',
how='inner'
)
# 创建以甲基化barcode为索引的Series
leiden_series = annotation_map.set_index('barcode')['rna_leiden']
# 将rna_leiden添加到adata_methy(只添加能匹配到的细胞)
adata_methy.obs['rna_leiden'] = leiden_series.reindex(adata_methy.obs.index)
elif reagent_type == "DD-MET5":
# DD-MET5: 直接通过barcode相等进行映射
# RNA和甲基化的barcode完全匹配,可以直接通过索引映射
adata_methy.obs['rna_leiden'] = adata_rna.obs.loc[adata_methy.obs.index, 'leiden'].values
else:
print(f"警告:未知的reagent_type: {reagent_type}")综合判定双细胞
综合判定策略:
通过整合 RNA 转录组聚类结果和甲基化 Reads 数信息,对双细胞进行联合判定:
RNA 转录组判定:基于 RNA 数据的 Leiden 聚类结果,识别具有异常标志基因共表达特征的细胞群。当某一聚类(如
cluster 9)中的细胞同时表达来自不同细胞类型的标志基因时,该聚类被定义为潜在的 RNA 双细胞群。甲基化 Reads 数判定:由于双细胞包含来自两个细胞的 DNA,其甲基化测序深度(Reads 数)通常显著高于单细胞,在 Reads 数排序分布中往往集中于高 Reads 数区间。
综合判定:将 RNA 转录组的 Leiden 聚类标签 (rna_leiden) 映射至甲基化数据。对于 rna_leiden 取特定值(如 9)的细胞,结合其在甲基化 Reads 数分布中的位置进行联合判定。
- 当细胞同时满足以下条件时,被最终认定为双细胞: 属于 RNA 层面判定的潜在双细胞聚类,在甲基化 Reads 数分布中位于高覆盖度区域。
# --- 标记rna_doublet:当rna_leiden为9时,标记为"doublet" ---
if 'rna_leiden' in adata_methy.obs.columns:
# 初始化rna_doublet列,默认值为空字符串或'singlet'
adata_methy.obs['rna_doublet'] = 'singlet'
# 当rna_leiden为9时(处理字符串或数字类型),标记为"rna_doublet"
adata_methy.obs.loc[adata_methy.obs['rna_leiden'].astype(str) == '9', 'rna_doublet'] = 'doublet'方法三:甲基化率鉴定双细胞
使用 MethylScrublet 算法来识别潜在的双细胞。该算法通过模拟人造双细胞,并将其与观测数据进行比对,从而计算每个细胞的"双细胞评分"。
# --- 打开 MCDS 格式的甲基化数据(1Mb 分辨率,用于 MethylScrublet) ---
# 根据新的文件层级结构构建路径:data/{top_dir}/methylation/{demo_dir}/{sample}/allcools_generate_datasets/{sample}.mcds
top_dir = sample_path_config[current_sample]['top_dir']
demo_dir = sample_path_config[current_sample]['demo_dir']
mcds_path = os.path.join('../../',data_root, top_dir, 'methylation', demo_dir, current_sample, 'allcools_generate_datasets', f'{current_sample}.mcds')
mcds = MCDS.open(
mcds_path,
obs_dim="cell",
var_dim="chrom1M"
)
# --- 提取甲基化计数(mc)和覆盖度(cov)矩阵 ---
mc = mcds[f'chrom1M_da'].sel({'count_type': 'mc'})
cov = mcds[f'chrom1M_da'].sel({'count_type': 'cov'})
# --- 如果数据量较小,加载到内存以提高计算速度 ---
load = True
if load and (mcds.get_index('cell').size <= 20000):
mc.load()
cov.load()# --- 初始化 MethylScrublet 对象 ---
scrublet = MethylScrublet(
sim_doublet_ratio=2.0, # 模拟双细胞数量 = 真实细胞数 × 2.0
n_neighbors=10, # KNN 邻居数
expected_doublet_rate=0.06, # 预期双细胞率
stdev_doublet_rate=0.02, # 双细胞率的标准差
metric='euclidean', # 距离度量方法
random_state=0, # 随机种子(保证结果可重复)
n_jobs=-1 # 使用所有可用 CPU 核心
)
# --- 运行双细胞识别算法 ---
# clusters=None 表示不使用先验的细胞类型信息
score, judge = scrublet.fit(mc, cov, clusters=None)
# --- 将结果保存到 AnnData 对象 ---
adata_methy.obs['met_doublet_score'] = score # 双细胞评分(0-1,越高越可能是双细胞)
adata_methy.obs['met_is_doublet'] = judge # 双细胞判定结果(True/False)
# --- 将判定结果转换为分类类型 ---
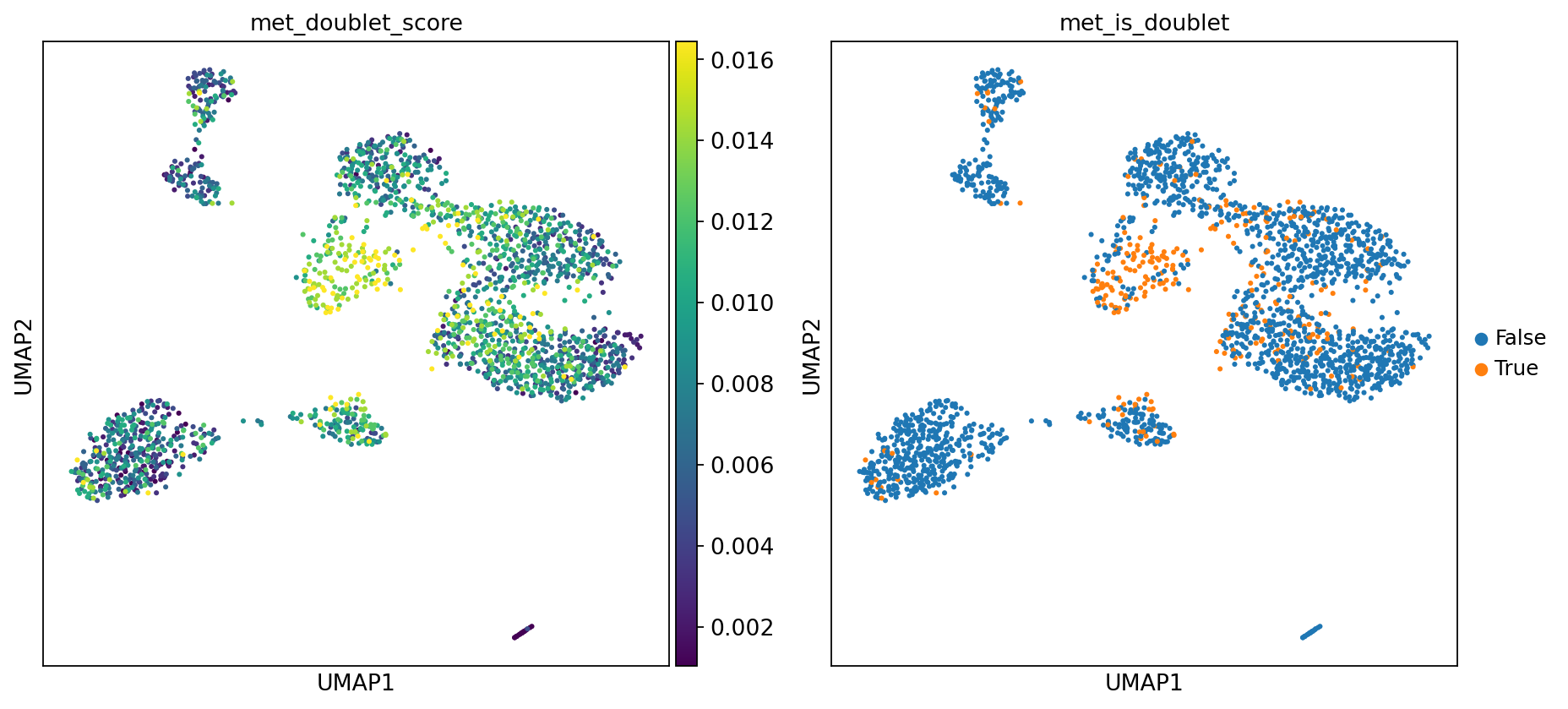
adata_methy.obs['met_is_doublet'] = adata_methy.obs['met_is_doublet'].astype('category')Detected doublet rate = 12.4%
Estimated detectable doublet fraction = 56.6%
Overall doublet rate:
Expected = 6.0%
Estimated = 21.9%
图注: 本图展示了 MethylScrublet 算法的双细胞识别结果,包括双细胞评分分布和最终判定结果。
- 左图 (met_doublet_score):双细胞评分分布。颜色越亮(黄色),表示该细胞与模拟的双细胞特征越相似,是双细胞的可能性越高。
- 右图 (met_is_doublet):双细胞判定结果。
- 橙色点 (True):被判定为双细胞 (Doublet)。
- 蓝色点 (False):判定为正常单细胞 (Singlet)。
warnings.filterwarnings('ignore')
# --- 设置图形参数 ---
sc.set_figure_params(scanpy=True, fontsize=12, facecolor='white', figsize=(6, 6))
# --- 在 UMAP 空间中可视化双细胞评分和判定结果 ---
sc.pl.umap(
adata_methy,
color=['met_doublet_score', 'met_is_doublet'],
ncols=2,
s=30
)
adata_methy.obs["m_cb"] = adata_methy.obs.index
# 输出双细胞判断结果文件:{sample}_doublet.txt
output_file = f"{current_sample}_doublet.txt"
# 保存列:m_cb, met_is_doublet, cell_multi_highlight, rna_doublet
adata_methy.obs[["m_cb","met_is_doublet","cell_multi_highlight","rna_doublet"]].to_csv(output_file, sep="\t", index=None)