甲基化 + RNA 双组学: MethSCAn 差异甲基化分析
模块简介
本模块基于 MethSCAn 开发,用于开展单细胞甲基化数据的系统性分析。
MethSCAn 是一套命令行工具集,支持单细胞甲基化数据的预处理、质量控制、变异区域识别及下游分析。该工具通过检测基因组范围内的甲基化变异,实现以下核心分析目标:
- 变异区域发现 (VMR Detection):自动识别细胞间甲基化变异区域(Variably Methylated Regions, VMRs),这些区域通常与细胞类型、发育阶段或疾病状态相关。
- 细胞聚类分群 (Cell Clustering):基于甲基化特征矩阵,对细胞进行聚类分群,有效区分不同的细胞类型或细胞状态,方法思路与单细胞 RNA 测序中的细胞聚类分析相似。
- 差异甲基化分析 (DMR Analysis):识别不同细胞群体之间的差异甲基化区域(Differentially Methylated Regions, DMRs),用于刻画表观遗传层面的细胞异质性。
输入文件准备
本模块支持从 compact_data 目录作为分析起点。compact_data 目录通常由以下数据预处理流程生成:
数据预处理流程
- 甲基化感知比对和提取:使用 allcools 分析流程进行比对和甲基化位点提取,生成包含甲基化和非甲基化位点信息的
allc文件。 - 格式转换:使用
allc_to_bismarkCov.py将allc文件转换为 bismark 的 coverage 格式(.cov文件)。 - 数据准备:使用
methscan prepare将.cov文件转换为高效的compact_data格式,供后续分析使用。
注意:
- 虽然 MethSCAn 支持
allc格式的输入,但其处理效率较低,通常不推荐在正式分析中直接使用,建议先转换为 bismark coverage 格式后再进行分析。 - 一般情况下,寻因云平台已默认完成
allc_to_bismarkCov.py和methscan prepare的步骤,可直接使用现有的compact_data目录开展分析。
文件结构示例
compact_data 目录结构如下:
compact_data/
├── chr1.npz
├── chr2.npz
├── chr3.npz
├── ...
├── chrX.npz
├── column_header.txt
└── cell_stats.csvMethSCAn 分析流程
脚本说明: 以下代码为 MethSCAn 分析流程的初始化示例,包含环境配置、路径设置及辅助函数定义,供后续各步骤调用。
options(warn = -1)
# 加载所需的 R 包
suppressPackageStartupMessages({
library(ggplot2) # 用于数据可视化
library(dplyr) # 用于数据处理
library(readr) # 用于读取 CSV 文件
library(tidyverse) # 数据整理和可视化工具集
library(data.table) # 高效的数据读取和处理
library(irlba) # 用于迭代 PCA(填补缺失值)
library(Seurat) # 单细胞分析工具(版本 v5)
library(Matrix) # 稀疏矩阵处理
})
# --- 输入参数配置 ---
## methscan_path:MethSCAn 命令路径
# 如果 methscan 在系统 PATH 中,设置为 NULL
# 如果 methscan 在不同环境中,指定完整路径
methscan_path <- "/jp_envs/envs/methscan/bin/methscan"
## outdir:结果输出目录
outdir <- "./DMRs"
# 创建输出目录(如不存在)
dir.create(outdir, recursive = TRUE, showWarnings = FALSE)
## compact_data_dir:compact_data 目录(由 methscan prepare 生成)
compact_data_dir <- file.path("../../data/AY1768874914782/methylation/demoWTJW969-task-1/WTJW969/methscan/compact_data")
## n_threads:并行计算线程数
# 注意:MethSCAn 会自动检测系统的最大线程数,当 n_threads 设置大于系统的最大线程数时会出错
# 建议设置为小于或等于系统可用 CPU 核心数
n_threads <- 8
# 定义函数:在 R 中调用 MethSCAn 命令
run_methscan <- function(command, args = character(), methscan_path = NULL) {
# 确定命令路径:优先使用参数,其次使用全局变量,最后使用系统 PATH
if (is.null(methscan_path) && exists("methscan_path", envir = .GlobalEnv)) {
methscan_path <- get("methscan_path", envir = .GlobalEnv)
}
cmd <- if (is.null(methscan_path)) "methscan" else methscan_path
cat("执行:", cmd, command, paste(args, collapse = " "), "\n")
# 创建临时文件用于捕获 stdout 和 stderr
stdout_file <- tempfile()
stderr_file <- tempfile()
# 执行命令,捕获输出
result <- system2(
cmd,
args = c(command, args),
wait = TRUE,
stdout = stdout_file,
stderr = stderr_file
)
return(invisible(result))
}关键参数设置
本节对 MethSCAn 分析流程中涉及的关键参数进行集中说明。这些参数将在后续的methscan filter、methscan scan、methscan matrix 和 methscan diff步骤中被调用,用于控制细胞质量过滤、变异区域检测及差异分析等关键行为。
methscan filter 参数
低质量细胞过滤相关的关键参数(如 min_sites、min_meth、max_meth 等)的具体含义及推荐取值,详见第 3.2.2 节中过滤执行代码前的参数说明表。
methscan diff 参数
用于比较不同 cluster 间差异的甲基化区域 (DMR) 分析所涉及的关键参数如下:
| 参数名称 | 中文释义 | 默认值 | 详细说明 |
|---|---|---|---|
min_cells | 最小细胞数要求 | 6 | 质量控制 (QC) 参数。 要求每个基因组区域在每个比较组中至少有该数量的细胞具有测序覆盖度,方可纳入差异分析。例如,设置为 6 表示仅对在每个组中至少有 6 个细胞具备覆盖度的区域进行差异检验。 注意:该参数用于过滤覆盖度不足的区域,有助于提高 DMR 检测的稳定性。如果某个组中细胞数量较少,可适当降低此值,但可能会增加假阳性风险。 |
threads 参数(通用参数)
用于控制计算密集型步骤(如 scan、matrix、diff)的并行计算线程数。
| 参数名称 | 中文释义 | 默认值/建议值 | 详细说明 |
|---|---|---|---|
threads | 并行计算线程数 | 8 | 性能优化参数。 用于加速计算密集型步骤(如 scan、matrix、diff),建议根据实际可用 CPU 核心数进行设置。重要提示:MethSCAn 会自动检测系统的最大线程数。当 threads 设置大于系统的最大线程数时,程序会报错。因此,建议将 threads 设置为小于或等于系统可用 CPU 核心数的值。 |
最佳实践建议
关于
methscan filter参数的选择:在参数设定前,强烈建议先运行质量评估步骤(见 3.2.1 节),通过可视化细胞的甲基化位点数和全局甲基化百分比分布,识别潜在离群细胞,并据此确定
min_sites、min_meth和max_meth的取值范围。min_sites参数策略:当过滤后保留的细胞数量较少时,可适当降低该阈值(例如降至 20000),但可能引入更多噪声;在数据质量较高的情况下,可适当提高阈值以增强结果可靠性。全局甲基化百分比阈值(
min_meth和max_meth):不同物种的全局甲基化水平通常存在差异,例如:- 小鼠:通常为 70-80%。
- 人类:通常为 60-70%。
关于计算资源:
methscan scan、matrix、diff:默认使用 8c64g 资源。在常见数据规模下,整个运行过程通常需要 1-2 小时。若在寻因云平台环境中运行,建议适当预留更长的任务时间窗口。关于 DMR 分析的
min_cells参数:默认值:每组 6 个细胞
参数作用:要求每个比较组中有足够数量的细胞在某一区域具有测序覆盖度,从而提高差异甲基化分析的统计稳定性。
经验参考:在实践中,当参与比较的两个 cluster 的细胞数量均大于约 200 个时,通常更容易获得满足显著性阈值的 DMR 结果;在细胞数量较少的情况下,可能难以检出显著差异,可尝试适当降低
min_cells参数进行探索性分析,并结合生物学背景对结果进行综合判断。
# --- 平台说明 ---
# 在寻因云平台环境中,methscan prepare 步骤通常已在上游流程中完成,
# 因此本流程默认直接使用已有的 compact_data 目录。
# 以下被注释的代码仅作为在本地环境或需要重新执行 prepare 步骤时的参考实现;
# 如需重新运行 methscan prepare,请取消相应注释。
# # 检查输入目录是否存在
# if (!dir.exists(input_cov_dir)) {
# stop("输入目录不存在:", input_cov_dir,
# "\n请先运行 allc_to_bismarkCov.py 转换数据")
# }
#
# # 获取所有 coverage 文件
# cov_files <- list.files(input_cov_dir, full.names = TRUE, pattern = "\\.cov$")
# if (length(cov_files) == 0) {
# stop("在 ", input_cov_dir, " 中未找到 .cov 文件")
# }
#
# cat("找到 ", length(cov_files), " 个 coverage 文件\n")
#
# # 准备输出目录
# compact_data_dir <- file.path(outdir, "compact_data")
# if (!dir.exists(compact_data_dir)) {
# dir.create(compact_data_dir, recursive = TRUE)
# }
#
# # 执行 methscan prepare
# run_methscan(
# command = "prepare",
# args = c(cov_files, compact_data_dir)
# )
# 直接使用已有的 compact_data 目录
if (!dir.exists(compact_data_dir)) {
stop("compact_data 目录不存在:", compact_data_dir,
"\n请确保已在云平台完成 methscan prepare 步骤")
}
cat("✓ 使用已有的 compact_data 目录:", compact_data_dir, "\n")质量评估和低质量细胞过滤
在执行细胞过滤之前,建议先对细胞质量进行系统评估,并结合可视化结果确定合适的过滤参数。
细胞统计信息读取与质量指标可视化
# 读取 cell_stats.csv 文件
cell_stats_path <- file.path(compact_data_dir, "cell_stats.csv")
cell_stats <- read_csv(cell_stats_path)
#if (file.exists(cell_stats_path)) {
# cell_stats <- read_csv(cell_stats_path)
# cat("成功读取细胞统计信息,共", nrow(cell_stats), "个细胞\n")
# head(cell_stats)
#} else {
# stop("找不到 cell_stats.csv 文件,请检查路径是否正确")
#}
# 可视化甲基化位点数和全局甲基化百分比
# 根据可视化结果,制定过滤参数
options(repr.plot.width = 8, repr.plot.height = 7, repr.plot.res = 150)
p <- cell_stats %>%
ggplot(aes(x = global_meth_frac * 100, y = n_obs)) +
geom_point(alpha = 0.6) +
# 在横坐标65和86处添加红色垂直线(示例阈值)
geom_vline(xintercept = c(65, 86), color = "red", linetype = "solid", linewidth = 1) +
# 在纵坐标60000处添加红色水平线(示例阈值)
geom_hline(yintercept = 60000, color = "red", linetype = "solid", linewidth = 1) +
labs(
x = "Global DNA methylation %",
y = "# of observed CpG sites",
title = "Cell Quality Assessment"
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, size = 14, face = "bold")
)
print(p)── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): cell_name
dbl (3): n_obs, n_meth, global_meth_frac
ℹ Use \`spec()\` to retrieve the full column specification for this data.
ℹ Specify the column types or set \`show_col_types = FALSE\` to quiet this message.
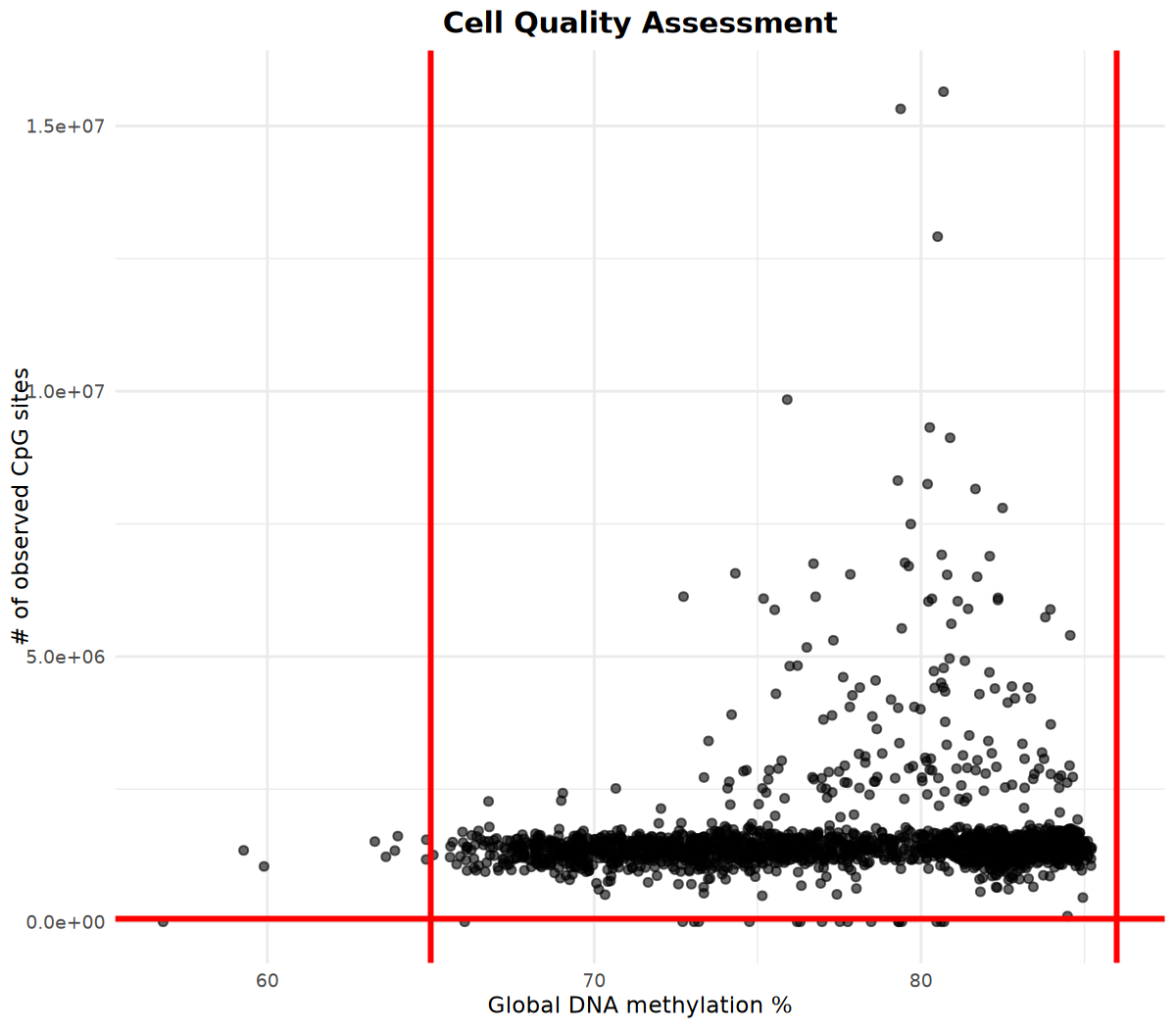
根据可视化结果执行过滤
基于上述可视化结果,确定过滤参数并执行细胞过滤:
methscan filter 关键参数:
| 参数名称 | 中文释义 | 默认值/建议值 | 详细说明 |
|---|---|---|---|
min_sites | 最小观察到的甲基化位点数 | 25000-60000 | 质量控制 (QC) 参数。 要求每个细胞观察到的甲基化位点数必须大于此值。用于剔除测序深度不足、无法准确推断甲基化状态的低质量细胞。 注意:此阈值取决于测序深度,深度越高,阈值可设置越高。 |
min_meth | 最小全局甲基化百分比 | 60-70 | 质量控制 (QC) 参数。 要求每个细胞的全局甲基化百分比必须大于此值。用于剔除可能由于不完全亚硫酸氢盐转化或技术问题导致的异常细胞。 注意:不同物种和细胞类型的正常甲基化水平差异很大(如小鼠通常为 70-80%,人类为 60-70%)。 |
max_meth | 最大全局甲基化百分比 | 70-85 | 质量控制 (QC) 参数。 要求每个细胞的全局甲基化百分比必须小于此值。用于剔除可能由于技术问题导致的异常高甲基化细胞。 注意:不同物种和细胞类型的正常甲基化水平差异很大,请根据实际数据情况设置合理的阈值范围。 |
threads | 并行计算线程数 | 8 | 用于加速计算密集型步骤(如 scan、matrix)。建议根据系统资源设置,通常设置为可用 CPU 核心数。 |
# 过滤参数(请根据质量评估结果调整)
min_sites <- 60000 # 最小观察到的甲基化位点数
min_meth <- 65 # 最小全局甲基化百分比
max_meth <- 86 # 最大全局甲基化百分比
# 准备输出目录
filtered_data_dir <- file.path(outdir, "filtered_data")
if (!dir.exists(filtered_data_dir)) {
dir.create(filtered_data_dir, recursive = TRUE)
}
# 执行 methscan filter
run_methscan(
command = "filter",
args = c(
"--min-sites", min_sites,
"--min-meth", min_meth,
"--max-meth", max_meth,
compact_data_dir,
filtered_data_dir
)
)数据平滑:methscan smooth
在进行 VMR/DMR 检测之前,需要对甲基化数据进行平滑处理。methscan smooth 将所有单细胞视为伪 bulk 样本,计算全基因组范围内的平滑平均甲基化水平。该结果是后续 VMR 检测及甲基化矩阵构建步骤的必要输入。
输出结果:平滑处理后的结果文件输出至 ${outdir}/filtered_data/smoothed 目录。
# 执行 methscan smooth
run_methscan(
command = "smooth",
args = filtered_data_dir
)VMR 检测:methscan scan
本步骤用于检测变异甲基化区域(Variable Methylation Region,VMR)。methscan scan 将在全基因组范围内自动识别细胞间甲基化变异显著的区域。
输出结果:生成 BED 格式的结果文件,包含 VMR 的基因组坐标和甲基化方差。
替代方案:若分析目标限定于特定的基因组区域(如启动子或基因体区域),可直接提供相应的 BED 文件,跳过 scan 步骤,直接使用 methscan matrix 分析这些区域。
# VMR 输出文件
vmr_bed_file <- file.path(outdir, "VMRs.bed")
# 执行 methscan scan
result <- run_methscan(
command = "scan",
args = c(
"--threads", n_threads,
filtered_data_dir,
vmr_bed_file
)
)VMR 矩阵生成:methscan matrix
本步骤基于已识别的 VMR 区域构建甲基化矩阵,用于后续的细胞聚类及相关下游分析。
输出文件说明:
输出目录 VMR_matrix中包含以下文件:
| 文件名 | 说明 |
|---|---|
mean_shrunken_residuals.csv.gz | 收缩残差矩阵(推荐用于下游分析)。受随机覆盖度变化影响较小,更适合用于降维和聚类分析。 |
methylation_fractions.csv.gz | 甲基化分数矩阵。每个区域的甲基化分数(0-1 之间),表示该区域的平均甲基化水平。 |
methylated_sites.csv.gz | 甲基化位点数矩阵(分子)。每个区域中甲基化的 CpG 位点数量。 |
total_sites.csv.gz | 总位点数矩阵(分母)。每个区域中具有测序覆盖度的 CpG 位点总数。 |
矩阵特征:
- 缺失值较多:受单细胞测序覆盖度较低的影响,部分基因组区域在部分细胞中可能不存在有效测序 reads,从而产生较多缺失值。
- 甲基化值呈小分母分数形式:甲基化水平通常以小分母分数表示(如 1/1、2/2、1/3 等),这是单细胞甲基化数据的典型特征。
- 附加位点统计信息:输出目录中同时包含每个区域的甲基化 CpG 位点数及具有测序覆盖的 CpG 位点数,分别对应甲基化比例计算中的分子和分母信息。
# VMR 矩阵输出目录
vmr_matrix_dir <- file.path(outdir, "VMR_matrix")
# 执行 methscan matrix
result <- run_methscan(
command = "matrix",
args = c(
"--threads", n_threads,
vmr_bed_file,
filtered_data_dir,
vmr_matrix_dir
)
)
# 检查输出文件是否生成
if (result == 0 && dir.exists(vmr_matrix_dir)) {
expected_files <- c(
"mean_shrunken_residuals.csv.gz",
"methylation_fractions.csv.gz",
"methylated_sites.csv.gz",
"total_sites.csv.gz"
)
existing_files <- list.files(vmr_matrix_dir)
found_files <- expected_files[expected_files %in% existing_files]
if (length(found_files) > 0) {
cat("✓ VMR 矩阵文件已生成:\n")
for (f in found_files) {
file_path <- file.path(vmr_matrix_dir, f)
file_size <- file.info(file_path)$size / 1024 / 1024 # MB
cat(" -", f, sprintf("(%.2f MB)\n", file_size))
}
} else {
warning("警告:未找到预期的输出文件,请检查输出目录:", vmr_matrix_dir)
}
} else if (result != 0) {
stop("methscan matrix 执行失败,请检查上面的错误信息")
}✓ VMR 矩阵文件已生成:
- mean_shrunken_residuals.csv.gz (291.34 MB)
- methylation_fractions.csv.gz (60.59 MB)
- methylated_sites.csv.gz (43.48 MB)
- total_sites.csv.gz (56.35 MB)
基于 VMR 的聚类分析
本节基于 VMR 甲基化矩阵,使用 Seurat 进行降维与聚类分析,以识别具有相似甲基化特征的细胞群体。
读取 VMR 矩阵
# 使用 fread 读取压缩文件
# 使用配置的 outdir 变量
vmr_matrix_path <- file.path(outdir, "VMR_matrix", "mean_shrunken_residuals.csv.gz")
if (file.exists(vmr_matrix_path)) {
cat("正在读取 VMR 矩阵...\n")
meth_mtx <- fread(vmr_matrix_path)
cat("矩阵维度:", nrow(meth_mtx), "行(细胞)x", ncol(meth_mtx) - 1, "列(VMR 区域)\n")
# 将第一列设置为行名并转换为矩阵
row_names <- meth_mtx[[1]]
meth_mtx <- as.matrix(meth_mtx[, -1])
rownames(meth_mtx) <- row_names
cat("矩阵转换完成!\n")
cat("缺失值比例:", round(mean(is.na(meth_mtx)) * 100, 2), "%\n")
} else {
stop("找不到 VMR 矩阵文件,请检查路径是否正确")
}矩阵转换完成!
缺失值比例: 75.31 %
迭代 PCA 填补缺失值
VMR 甲基化矩阵在结构上与单细胞 RNA-seq 的 count 矩阵类似,但其显著特点是包含大量缺失值。这主要源于单细胞甲基化测序覆盖度较低,导致许多基因组区域在部分细胞中未获得有效测序 reads。
为处理上述缺失问题,本分析采用迭代 PCA 方法对缺失值进行填补。具体过程为:首先将缺失值以 0 进行初始化填充;随后在填补后的矩阵上执行 PCA,并使用 PCA 重构得到的预测值替换原先填入的 0;该过程反复迭代,直至填补值在相邻迭代间变化趋于稳定。
该方法通过迭代优化低维结构,对缺失甲基化值进行更合理的估计,从而为后续的降维和聚类分析提供更完整、稳定的数据基础。
# 定义迭代 PCA 填补缺失值的函数
prcomp_iterative <- function(x, n = 15, n_iter = 50, min_gain = 0.001, ...) {
mse <- rep(NA, n_iter)
na_loc <- is.na(x)
x[na_loc] <- 0 # zero is our first guess
for (i in 1:n_iter) {
prev_imp <- x[na_loc] # what we imputed in the previous round
# PCA on the imputed matrix
pr <- prcomp_irlba(x, center = F, scale. = F, n = n, ...)
# impute missing values with PCA
new_imp <- (pr$x %*% t(pr$rotation))[na_loc]
x[na_loc] <- new_imp
# compare our new imputed values to the ones from the previous round
mse[i] <- mean((prev_imp - new_imp) ^ 2)
# if the values didn't change a lot, terminate the iteration
gain <- mse[i] / max(mse, na.rm = T)
if (gain < min_gain) {
message(paste(c("\n\nTerminated after ", i, " iterations.")))
break
}
}
message(paste(c("\n\nTerminated after ", i, " iterations. Gain: ", round(gain, 6))))
pr$mse_iter <- mse[1:i]
list(pr = pr, imputed_matrix = x)
}
cat("开始执行迭代 PCA 填补缺失值...\n")
result_residual <- meth_mtx %>%
scale(center = T, scale = F) %>%
prcomp_iterative(n = 15)
# 提取填补后的残差矩阵
residual_mtx_imputed <- result_residual$imputed_matrix
cat("缺失值填补完成!\n")Terminated after 32 iterations.
Terminated after 32 iterations. Gain: 0.000974
缺失值填补完成!
创建 Seurat 对象
使用经缺失值填补后的残差矩阵(residual_mtx_imputed)作为 Seurat 对象的输入。在构建 Seurat 对象时,counts、data 和 scale.data 三个矩阵均使用相同的填补后残差矩阵。
说明:后续的 PCA、降维和聚类分析实际基于 scale.data 矩阵进行。
# 使用填补缺失值后的残差矩阵创建 Seurat 对象
# counts、data 和 scale.data 三个矩阵使用相同的填补后的残差矩阵
seurat_obj <- CreateSeuratObject(
counts = as(t(residual_mtx_imputed), "sparseMatrix"),
project = "MethSCAn",
assay = "VMR"
)
# 将 data 和 scale.data 设置为与 counts 相同的矩阵
# 注意:后续的 PCA、降维和聚类分析实际使用的是 scale.data 矩阵
seurat_obj@assays$VMR@data <- seurat_obj@assays$VMR@counts
seurat_obj@assays$VMR@scale.data <- as.matrix(seurat_obj@assays$VMR@counts)PCA、UMAP 降维和聚类
# 1. PCA 降维
# 使用 Seurat 的 RunPCA 函数
# 这会自动从 scale.data 层读取数据并计算 PCA
n_pcs = 30
seurat_obj <- RunPCA(
seurat_obj,
features = rownames(seurat_obj), # 使用所有 VMRs
npcs = n_pcs, # 计算前 30 个主成分
assay = "VMR",
verbose = FALSE
)
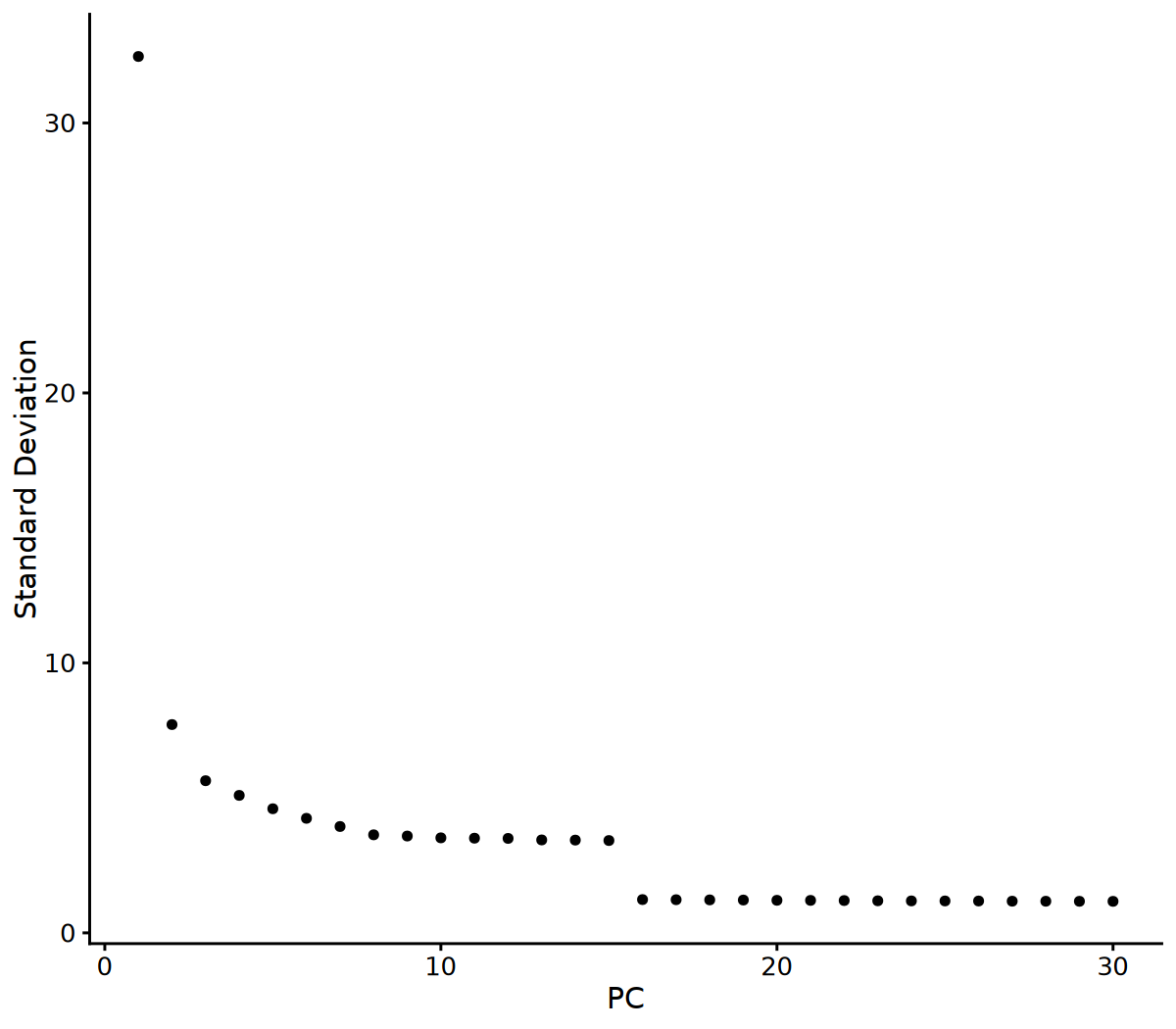
options(repr.plot.width = 8, repr.plot.height = 7, repr.plot.res = 150)
# 可视化 Elbow Plot 以确定使用的主成分数量
ElbowPlot(seurat_obj, ndims = 30)
# 2. 根据 Elbow Plot 确定使用的主成分数量
# 您可以根据 Elbow Plot 的结果调整这个参数
n_pcs_use <- 10
# 3. UMAP 降维
seurat_obj <- RunUMAP(seurat_obj, dims = 1:n_pcs_use, reduction = "pca", verbose = FALSE)
cat("UMAP 降维完成!\n")
# 4. 构建 KNN 图
seurat_obj <- FindNeighbors(seurat_obj, dims = 1:n_pcs_use, reduction = "pca", verbose = FALSE)
cat("KNN 图构建完成!\n")
# 5. 聚类(resolution 参数可以调整,值越大,聚类数越多)
seurat_obj <- FindClusters(seurat_obj, resolution = 0.5, verbose = FALSE)
cat("聚类完成!\n")
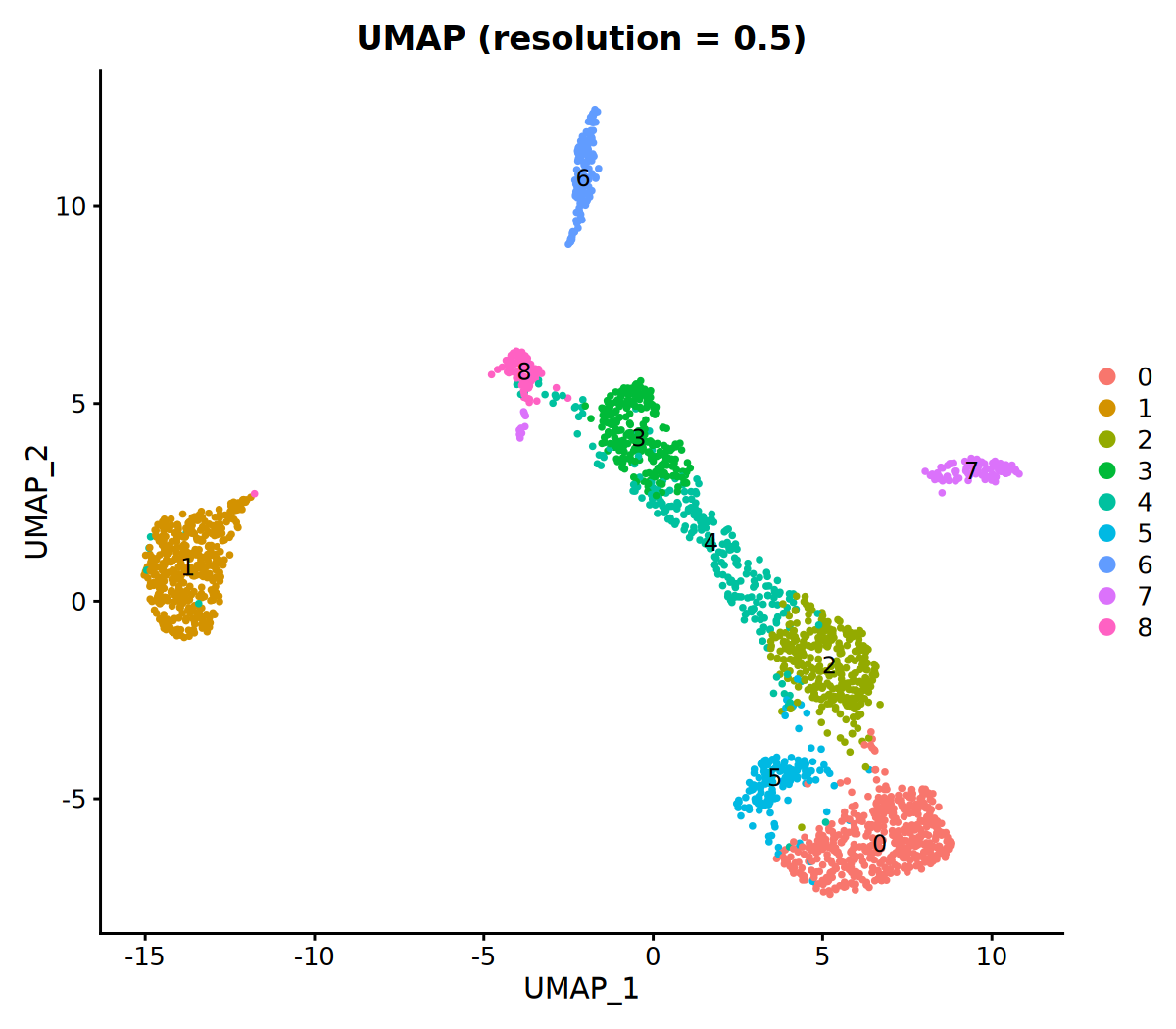
# 6. 可视化聚类结果
DimPlot(seurat_obj, group.by = 'VMR_snn_res.0.5', label = TRUE) +
ggtitle("UMAP (resolution = 0.5)")KNN 图构建完成!
聚类完成!
保存结果
# 保存 Seurat 对象
output_file <- "MethSCAn.rds"
saveRDS(seurat_obj, file = output_file)clust_tbl <- seurat_obj@meta.data
clust_tbl$cell <- row.names(clust_tbl)
clust_tbl %>%
mutate(cell_group = case_when(
seurat_clusters == "0" ~ "-",
seurat_clusters == "1" ~ "1",
seurat_clusters == "2" ~ "2",
seurat_clusters == "3" ~ "-",
seurat_clusters == "4" ~ "-",
seurat_clusters == "5" ~ "-",
seurat_clusters == "6" ~ "-",
seurat_clusters == "7" ~ "-",
seurat_clusters == "8" ~ "-"
)) %>%
dplyr::select(cell, cell_group) %>%
write_csv("cell_groups.csv", col_names=F)差异甲基化区域分析:methscan diff
差异甲基化区域 (DMR) 分析用于比较不同条件下细胞之间的甲基化差异,例如不同 cluster、不同细胞类型或其他自定义分组。通常建议在完成聚类分析后,基于聚类结果或预定义分组开展 DMR 分析。
输入文件准备
输入要求:
- 数据目录:
filtered_data目录(需已完成methscan smooth步骤)。 - 细胞分组文件 (cell_groups_file):CSV 格式文件,其中第一列为细胞名称 (cell barcode),第二列为分组标签。标签规则为:
group_A和group_B表示参与比较的两个细胞组;-表示不参与本次比较的其他细胞。
文件格式示例:
cell_01,-
cell_02,-
cell_03,-
cell_04,group_A
cell_05,group_A
cell_06,group_A
cell_07,group_B
cell_08,group_B
cell_09,group_B# 细胞分组文件(用于 DMR 分析)
cell_groups_file <- "cell_groups.csv" # 请根据实际情况修改
# DMR 输出文件
dmr_bed_file <- file.path(outdir, "DMRs.bed")
# 检查细胞分组文件是否存在
if (file.exists(cell_groups_file)) {
# 执行 methscan diff
run_methscan(
command = "diff",
args = c(
"--threads", n_threads,
filtered_data_dir,
cell_groups_file,
dmr_bed_file
)
)
} else {
warning("细胞分组文件不存在:", cell_groups_file,
"\n跳过 DMR 分析。如需进行 DMR 分析,请先准备细胞分组文件。")
}输出文件格式说明
输出文件:DMRs.bed
结果以 BED 格式输出,包含以下字段:
| 列号 | 列名 | 说明 | 示例值 |
|---|---|---|---|
| 1 | chromosome | 染色体名称 | 17 |
| 2 | DMR_start | DMR 起始位置(基因组坐标) | 58270529 |
| 3 | DMR_end | DMR 结束位置(基因组坐标) | 58285529 |
| 4 | t_statistic | Welch's t 检验统计量 | -12.85232790631932 |
| 5 | n_sites | 该区域内的甲基化位点总数(CpG 数量) | 561 |
| 6 | n_cells_group1 | 组 1 中有覆盖度的细胞数量 | 150 |
| 7 | n_cells_group2 | 组 2 中有覆盖度的细胞数量 | 66 |
| 8 | meth_frac_group1 | 组 1 的平均甲基化水平(0–1) | 0.3396857458943857 |
| 9 | meth_frac_group2 | 组 2 的平均甲基化水平(0–1) | 0.8429240842342447 |
| 10 | low_group_label | 甲基化水平较低的组标签 | group_1 |
| 11 | p | 原始 p 值(未调整) | 0.0 |
| 12 | adjusted_p | 调整后的 p 值(FDR,Benjamini-Hochberg 方法) | 0.0 |
结果解读:low_group_label 列表示甲基化水平较低的组标签;adjusted_p 列为多重检验校正后的显著性水平,通常建议以adjusted_p < 0.05作为筛选阈值;t_statistic 的符号可判断差异方向(负值表示 group_1 甲基化水平较低,正值表示 group_1 甲基化水平较高)。
生成显著差异甲基化区域文件
基于 DMRs.bed 文件,对显著差异甲基化区域进行筛选(adjusted_p < 0.05),并根据 low_group_label 对结果进行分组,生成两个 BED 文件。
输入: DMRs.bed:DMR 检测结果文件
输出:根据 low_group_label 的值,生成以下两个显著 DMR BED 文件:
DMRs_significant_group_A.bed:在 group_A 中甲基化水平显著较低的 DMR 区域。DMRs_significant_group_B.bed:在 group_B 中甲基化水平显著较低的 DMR 区域。
上述输出文件均为 BED 格式,包含三列:chromosome、start、end。其中,若染色体名称为数字或性染色体(X/Y),将自动添加 "chr" 前缀;若原本已包含 "chr" 前缀,则保持不变。
说明:仅保留 adjusted_p < 0.05 的显著差异甲基化区域,自动为数字染色体和性染色体添加 "chr" 前缀(若已有则保持不变),并跳过 low_group_label 为空或为 "-" 的行。
# 生成显著的 DMR 文件(按 low_group_label 分组,adjusted_p < 0.05)
# 在常染色体(数字)和性染色体(XY)前加上 "chr" 前缀
# 检查 DMRs.bed 文件是否存在
if (file.exists(dmr_bed_file)) {
cat("开始生成显著的 DMR 文件...\n")
# 构建 awk 命令
awk_script <- sprintf('
BEGIN {
outdir = "%s"
}
# 跳过表头或空行
NR == 1 && $1 ~ /^chromosome|^#/ { next }
NF < 12 { next }
# 筛选 adjusted_p < 0.05 的行
$12 < 0.05 {
# 获取 low_group_label(第10列)作为文件名
group_label = $10
if (group_label == "" || group_label == "-") next
# 处理染色体名称:如果是数字或 X/Y,则加上 "chr" 前缀
chrom = $1
if (chrom ~ /^[0-9]+$/ || chrom ~ /^[XY]$/) {
chrom = "chr" chrom
}
# 构建输出文件名
output_file = outdir "/DMRs_significant_" group_label ".bed"
# 输出前3列(chromosome, start, end)到对应的文件
print chrom "\\t" $2 "\\t" $3 > output_file
}
END {
print "处理完成!已生成显著的 DMR 文件。"
}
', outdir)
# 在 R 中执行 awk 命令
awk_cmd <- paste("awk -F'\\t'", shQuote(awk_script), shQuote(dmr_bed_file))
result <- system(awk_cmd, intern = TRUE)
# 显示输出
cat(paste(result, collapse = "\n"), "\n")
# 检查生成的文件
significant_files <- list.files(
path = outdir,
pattern = "^DMRs_significant_.*\\.bed$",
full.names = TRUE
)
if (length(significant_files) > 0) {
cat("\n生成的显著 DMR 文件:\n")
for (f in significant_files) {
n_lines <- length(readLines(f, warn = FALSE))
cat(" -", basename(f), "(", n_lines, " 个区域)\n", sep = "")
}
} else {
warning("未找到生成的显著 DMR 文件")
}
} else {
warning("DMRs.bed 文件不存在:", dmr_bed_file,
"\n请先执行 methscan diff 命令")
}生成的显著 DMR 文件:
-DMRs_significant_1.bed(23951 个区域)
-DMRs_significant_2.bed(30189 个区域)