SeekSpace 空间转录组多样本整合与批次效应评估
输入文件准备
本模块需要以下输入文件:
必需文件
- Seurat 对象文件:已释放数据的 Seurat 对象(
.rds格式),包含基因表达矩阵和细胞元数据。
文件结构示例
./
├── AA1.rds # 样本 1 的 Seurat 对象
├── AA3.rds # 样本 2 的 Seurat 对象
└── ...重要提示
输入的 Seurat 对象应包含以下信息:
- 基因表达矩阵(counts 或 data slot)
- 细胞元数据(meta.data),包括样本标识(
orig.ident)- 细胞空间位置信息
⚠️ 拼片处理提醒
在分析开始前,若样本存在拼片(tile 拼接),需要由相关生信人员拆分拼片后释放可分析数据 rds。请确保输入的 Seurat 对象文件已经完成拼片拆分处理。
# --- 导入必要的 R 包 ---
library(Seurat)
library(harmony)
library(dplyr)
library(tibble)
library(tidyverse)
library(ggplot2)Loading required package: Rcpp
Warning message:
“package ‘Rcpp’ was built under R version 4.3.3”
Warning message:
“package ‘dplyr’ was built under R version 4.3.3”
Attaching package: ‘dplyr’
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
Warning message:
“package ‘tibble’ was built under R version 4.3.3”
数据读取与整合
本节读取多个样本的 Seurat 对象文件,并进行数据合并。
数据读取流程
主要步骤:
定义文件列表:指定需要读取的 Seurat 对象文件(
.rds格式),这些文件应包含已释放的空间转录组数据。循环读取数据:
- 使用
readRDS()函数逐个读取每个样本的 Seurat 对象 - 提取并保存每个样本的元信息(
@misc$info),用于后续分析
- 使用
数据合并:
- 如果只有一个样本,直接使用该样本的数据
- 如果有多个样本,使用
merge()函数合并所有样本 - 使用
merge.dr = "spatial"参数确保空间维度信息(spatial dimension reduction)被正确合并 - 如果细胞名称存在重复,Seurat 会自动重命名以确保唯一性
信息保留:将各样本的元信息合并到整合后的 Seurat 对象中
注意事项:
- 确保所有
.rds文件位于当前工作目录或指定路径 - 合并后的数据将包含所有样本的细胞和基因信息
- 合并过程会自动处理细胞名称冲突问题
# ============================================================================
# 数据读取与合并
# ============================================================================
# --- 步骤 1: 定义需要读取的 Seurat 对象文件列表 ---
# 将需要整合分析的样本文件名添加到列表中
# 注意:请根据实际分析需求修改文件名
# 教程示例MIA6,MIA7两个样本
rds_files <- c("MIA6.rds", "MIA7.rds")
# --- 步骤 2: 初始化存储列表 ---
# data_list: 用于存储读取的各个样本的 Seurat 对象
# info2keep: 用于存储各个样本的元信息(@misc$info)
data_list <- list()
info2keep <- list()
# --- 步骤 3: 循环读取所有样本的 Seurat 对象文件 ---
# 遍历文件列表,逐个读取并提取信息
for (i in seq_along(rds_files)) {
# 显示当前正在读取的文件名
cat("正在读取:", rds_files[i], "\n")
# 使用 readRDS() 读取 Seurat 对象文件
data_list[[i]] <- readRDS(rds_files[i])
# 检查并提取样本的元信息(如果存在)
# @misc$info 通常包含样本的额外注释信息
if (length(data_list[[i]]@misc$info) > 0) {
info2keep <- append(info2keep, data_list[[i]]@misc$info)
}
}
# --- 步骤 4: 合并多个样本的数据 ---
# 根据样本数量选择合并策略
if (length(data_list) == 1) {
# 如果只有一个样本,直接使用该样本的数据
data <- data_list[[1]]
} else {
# 如果有多个样本,使用 merge() 函数合并
# merge.dr = "spatial": 指定合并空间维度信息(spatial dimension reduction)
# 这对于空间转录组数据非常重要,确保空间坐标信息被正确保留
# 注意:如果细胞名称重复,Seurat 会自动重命名以确保唯一性
data <- merge(data_list[[1]], y = data_list[-1], merge.dr = "spatial")
}
# --- 步骤 5: 保留样本元信息 ---
# 将各样本的元信息合并到整合后的 Seurat 对象中
if (length(info2keep) > 0) {
data@misc$info <- info2keep
}
# --- 步骤 6: 输出合并结果统计 ---
# 显示合并后的总细胞数,用于验证合并是否成功
cat("合并后细胞数:", ncol(data), "\n")
cat("合并后基因数:", nrow(data), "\n")正在读取: MIA7.rds
Warning message in CheckDuplicateCellNames(object.list = objects):
“Some cell names are duplicated across objects provided. Renaming to enforce unique cell names.”
合并后细胞数: 116353
数据质控与过滤
本节对整合后的数据进行质控指标计算和可视化,然后根据质控标准过滤低质量细胞。
质控指标计算
关键质控指标:
- nCount_RNA(总 UMI 数):每个细胞检测到的总 UMI 数,反映测序深度。
- nFeature_RNA(检测到的基因数):每个细胞检测到的基因数量,反映细胞捕获效率。
- percent.mt(线粒体基因比例):线粒体基因表达占比,用于识别低质量或死亡细胞。
质控过滤策略(以结果为导向,无绝对标准):
- 线粒体基因比例:过滤
percent.mt > 20%的细胞。线粒体基因比例过高的细胞可能已死亡或受损。 - 总 UMI 数:过滤
nCount_RNA < 200或nCount_RNA > 100000的细胞。UMI 数过少表示测序深度不足,过多可能是双细胞或技术异常。 - 检测到的基因数:过滤
nFeature_RNA < 1或nFeature_RNA > 10000的细胞。基因数过少表示捕获效率低,过多可能是双细胞。
图注说明(质控指标小提琴图):
下图展示了质控过滤前各质控指标的分布情况,按样本分组显示。
- 横坐标:不同样本,便于比较不同样本间的质控指标差异。
- 纵坐标:各质控指标的数值,包括
nCount_RNA(总 UMI 数)、nFeature_RNA(检测到的基因数)和percent.mt(线粒体基因比例)。- 小提琴图:每个小提琴图代表一个样本,宽度表示该数值区间的细胞密度,越宽表示该区间的细胞越多。
- 用途:用于评估原始数据质量,识别异常值,以及确定质控过滤阈值。
# --- 计算线粒体基因比例 ---
data[["percent.mito"]] <- PercentageFeatureSet(data, pattern = "^mt-")
# --- 质控指标小提琴图可视化 ---
VlnPlot(data, features = c("nCount_RNA", "nFeature_RNA","percent.mito"),group.by = "Sample",
ncol = 3,pt.size = 0)To disable this behavior set \`raster=FALSE\`
Warning message:
“\`aes_string()\` was deprecated in ggplot2 3.0.0.
ℹ Please use tidy evaluation idioms with \`aes()\`.
ℹ See also \`vignette("ggplot2-in-packages")\` for more information.
ℹ The deprecated feature was likely used in the Seurat package.
Please report the issue at
质控过滤
根据质控标准过滤低质量细胞。
# --- 质控过滤:去除低质量细胞 ---
data <- subset(data, subset = percent.mito < 20 &
nCount_RNA > 200 & nCount_RNA < 100000 &
nFeature_RNA > 1 & nFeature_RNA < 10000)
# 输出过滤后的细胞数
cat("过滤后细胞数:", ncol(data), "\n")
# --- 质控后小提琴图可视化 ---
VlnPlot(data, features = c("nCount_RNA", "nFeature_RNA", "percent.mito"), group.by = "Sample",
ncol = 3, pt.size = 0)Rasterizing points since number of points exceeds 100,000.
To disable this behavior set \`raster=FALSE\`
数据预处理与降维
本节对质控后的数据进行标准化、高变基因筛选、数据缩放和主成分分析(PCA)。
数据标准化与高变基因筛选
数据标准化 (NormalizeData):
- 功能:对原始 UMI 计数数据进行标准化处理,消除细胞间测序深度差异。
- 方法:采用对数标准化方法(
LogNormalize),基准测序深度为 10,000 个转录本。 - 公式:
log(1 + UMI_count / 10000)
高变基因筛选 (FindVariableFeatures):
- 功能:识别细胞间表达变异最显著的基因,聚焦具有生物学意义的特征基因。
- 方法:使用方差稳定变换法(
vst),选取前 2,000 个高变基因。 - 意义:高变基因通常对应细胞类型标记基因,排除技术噪音,保留真实生物学变异。
数据缩放 (ScaleData):
- 功能:对表达矩阵进行 Z-score 标准化,为降维分析准备标准化数据。
- 特征:每个基因的均值为 0,标准差为 1,消除基因间表达量级差异。
- 意义:避免高表达基因主导降维结果,确保所有基因在 PCA 中平等贡献。
主成分分析 (PCA)
功能概述:
- 对标准化后的高变基因进行主成分分析,提取数据的主要变异方向。
- PCA 将高维基因表达数据降维到低维空间,保留主要生物学信号。
- 前几个主成分通常捕获了细胞类型间的差异。
# --- 数据标准化 ---
data <- NormalizeData(data, normalization.method = "LogNormalize", scale.factor = 10000)
# --- 筛选高变基因 ---
data <- FindVariableFeatures(data, selection.method = "vst", nfeatures = 2000)
# --- 数据缩放(Z-score 标准化)---
# 注意:vars.to.regress 参数可用于回归掉特定变量(如线粒体基因比例、细胞周期等)
data <- ScaleData(data, features = VariableFeatures(data))
# --- 主成分分析 (PCA) ---
data <- RunPCA(data, features = VariableFeatures(object = data), reduction.name = "pca")PC_ 1
Positive: CACNA1C, LAMA2, ROBO2, PDZRN3, COL6A3, LTBP2, PIEZO2, ABI3BP, CDH11, RUNX1T1
LSAMP, ROBO1, NAV3, MEIS1, CALD1, ELN, A2M, LAMA4, ITGBL1, ADAMTS12
SGIP1, DST, ZFPM2, NR2F2-AS1, MIR100HG, UACA, PRDM6, FAT4, UNC5C, SLIT3
Negative: PLCH1, TMEM163, MUC1, LMO3, CADM1, CACNA2D2, STEAP4, KCNJ15, SCTR, CIT
GPR39, CDH1, SHROOM4, LHFPL3-AS2, HOPX, VEPH1, LRRK2, MEGF11, SORCS2, TOX
AC010998.1, MAGI3, ARHGEF2, GALNT10, GPRC5A, TMEM164, AC019117.1, C4orf19, SNX25, ESYT3
PC_ 2
Positive: CADPS2, PLXNA2, LMO3, ZNF385B, PLCH1, STEAP4, CADM1, MAGI3, PDE4D, MACROD2
SHROOM4, CDC42BPA, CACNA2D2, CACHD1, KCNJ15, DLG2, SORCS2, C1orf21, DLGAP1, SCTR
GLIS3, TMEM163, MUC1, PTPRD, LHFPL3-AS2, MEGF11, PRKG1, PID1, SDK1, VEPH1
Negative: WDFY4, SFMBT2, ALOX5, PDE4B, SAMSN1, ITGAX, LAPTM5, THEMIS2, KLHL6, ITGB2
CYBB, TBXAS1, AOAH, MRC1, SPI1, VAV3, KYNU, RUNX3, IFNG-AS1, HCK
IL7R, CSF1R, LINC01934, TFEC, MSR1, GRAMD1B, SIGLEC1, OLR1, NABP1, TRPM2
PC_ 3
Positive: LINC01876, C4BPA, AC104041.1, TP63, SOX5, WIF1, DNAH11, TOX, FOXP2, IGF2BP2
TTN, CTXND1, HMGA2, AC018742.1, SFTPC, IFNG-AS1, LINC01934, GLIS3, GPC6, ITGB6
PKHD1, IL7R, ERBB4, MEGF11, CACNB4, TESC, TNC, MTCL1, PTPRD, RYR2
Negative: MRC1, ITGAX, MSR1, SLCO2B1, NPL, FMNL2, OLR1, TNFAIP2, KCNMA1, GLUL
CTSB, SIGLEC1, DMXL2, PSAP, CLEC7A, SLC8A1, ADAMTSL4, NCF2, PLAUR, RAB20
CSF1R, HCK, MS4A7, NFAM1, PLXDC2, GPNMB, TGFBI, RASSF4, SPI1, SLC11A1
PC_ 4
Positive: KAZN, SFTPA2, CEACAM6, SFTPA1, GPC5, LGR4, AC046195.1, FSTL4, SLC22A31, NRXN3
MMP28, AC002070.1, CIT, LINC02257, CAPN9, LRRC36, MID1, NOS1AP, TOX3, AC027288.3
PLS3, ADGRF1, SLC35F3, GALNT17, CHST3, ARHGEF2, PTCHD1-AS, ABCA4, CDC42BPA, HMCN1
Negative: TP63, LINC01876, AC104041.1, C4BPA, WIF1, IGF2BP2, DNAH11, FOXP2, GLIS3, AC018742.1
CTXND1, MTCL1, CACNB4, MEGF11, ITGB6, MCTP1, HMGA2, NAMPT, TNC, SOX5
ERBB4, STK32B, LINC00923, SLC7A2, PTPRD, PKHD1, GPC6, FREM2, MACROD2, SFTPC
PC_ 5
Positive: LAMA2, LRP1, LSAMP, PLEKHH2, COL6A3, PDZRN3, CCBE1, SLIT2, ITGBL1, ROBO2
SCN7A, LTBP2, GLI2, ABCA6, FBLN1, PRDM6, RYR2, NKD2, ADAMTS12, MIR100HG
SPON1, SLIT3, CDH11, ANTXR1, BMP5, NCAM2, ANGPT1, MAMDC2, BICC1, ZEB2
Negative: VWF, SHANK3, PTPRB, EGFL7, ANO2, DIPK2B, AC000050.1, PCDH17, CALCRL, CYYR1
EMCN, TEK, LDB2, MYRIP, RAPGEF4, NOSTRIN, AL021937.3, ADGRL4, ROBO4, TLL1
SEMA6A, FLT1, NOTCH4, TIE1, CDH5, RAMP3, PALMD, JAM2, PCAT19, SNTG2
批次校正与降维可视化
本节使用 Harmony 算法校正批次效应,然后进行 UMAP 降维可视化、构建邻域图和细胞聚类。
批次校正 (Harmony)
功能概述:
- 消除不同样本/实验批次的技术差异,整合多来源数据,保留生物学变异。
- Harmony 是一种基于迭代优化的批次校正方法,能够有效校正批次效应,同时保留生物学信号。
参数说明:
reduction = "pca":基于 PCA 降维结果进行校正。group.by.vars = "orig.ident":按样本来源(orig.ident)分组校正。reduction.save = "harmony":保存校正后的嵌入结果,命名为 "harmony"。
应用价值:
- 解决批次效应,避免技术差异误导聚类。
- 使不同样本的相同细胞类型能够正确聚集。
降维可视化 (UMAP)
功能概述:
- 将高维数据投影到 2D 空间进行可视化,保留数据拓扑结构,展示细胞关系。
参数说明:
reduction = "harmony":使用 Harmony 校正后的 PCA 结果。dims = 1:30:使用前 30 个主成分。reduction.name = "umap":保存为 "umap" 降维结果。
可视化意义:
- 每个点代表一个细胞。
- 距离近的细胞表达谱相似。
- 直观展示细胞群体结构。
构建邻域图与细胞聚类
构建邻域图 (FindNeighbors):
- 功能:基于降维空间计算细胞间的相似性,构建 K 近邻图用于后续聚类分析。
- 参数:使用校正后的 PCA 结果(
reduction = "harmony"),使用前 30 个主成分(dims = 1:30)。 - 算法:计算细胞在降维空间中的欧氏距离,构建共享最近邻(SNN)图。
细胞聚类 (FindClusters):
- 功能:基于邻域图进行社区发现算法聚类,识别不同的细胞群体/亚群。
- 参数:
resolution = 1为聚类分辨率参数。 - 分辨率调节:
resolution = 0.4-0.8:较少、更大的聚类(粗聚类)。resolution = 1-2:较多、更小的聚类(细聚类)。- 可根据细胞类型复杂度调整。
# --- 批次校正 (Harmony) ---
data <- RunHarmony(data, reduction = "pca",
group.by.vars = "orig.ident",
reduction.save = "harmony")
# --- UMAP 降维可视化 ---
data <- RunUMAP(data, reduction = "harmony", dims = 1:30, reduction.name = "umap")
# --- 构建邻域图 ---
data <- FindNeighbors(data, reduction = "harmony", dims = 1:30)
# --- 细胞聚类 ---
data <- FindClusters(data, resolution = 1)Initializing state using k-means centroids initialization
Warning message:
“Quick-TRANSfer stage steps exceeded maximum (= 5815150)”
Warning message:
“Quick-TRANSfer stage steps exceeded maximum (= 5815150)”
Harmony 1/10
Harmony 2/10
Harmony converged after 2 iterations
Warning message:
“Invalid name supplied, making object name syntactically valid. New object name is Seurat..ProjectDim.RNA.harmony; see ?make.names for more details on syntax validity”
Warning message:
“The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
This message will be shown once per session”
17:23:06 UMAP embedding parameters a = 0.9922 b = 1.112
17:23:06 Read 116303 rows and found 30 numeric columns
17:23:06 Using Annoy for neighbor search, n_neighbors = 30
17:23:06 Building Annoy index with metric = cosine, n_trees = 50
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
|
17:23:18 Writing NN index file to temp file /tmp/RtmpUqWHQp/file7dd262b449d
17:23:18 Searching Annoy index using 1 thread, search_k = 3000
17:24:08 Annoy recall = 100%
17:24:09 Commencing smooth kNN distance calibration using 1 thread
with target n_neighbors = 30
17:24:13 Initializing from normalized Laplacian + noise (using irlba)
17:24:29 Commencing optimization for 200 epochs, with 5483780 positive edges
17:25:31 Optimization finished
Computing nearest neighbor graph
Computing SNN
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 116303
Number of edges: 3727964
Running Louvain algorithm...n Maximum modularity in 10 random starts: 0.8978
Number of communities: 35
Elapsed time: 52 seconds
1 singletons identified. 34 final clusters.
结果可视化
数据整合分析完成后,我们需要通过多种可视化方法来展示分析结果。本节将介绍三种主要的可视化方式:
- UMAP 降维可视化:展示细胞在降维空间中的分布,便于观察不同样本和细胞群体的关系。
- 空间位置可视化:展示细胞在组织样本中的实际空间位置,用于分析空间分布模式。
- 自定义空间绘图函数:提供更灵活的可视化方式,可以结合组织图像背景进行展示。
UMAP 降维可视化
功能概述:
UMAP(Uniform Manifold Approximation and Projection)降维可视化是单细胞和空间转录组分析中最常用的可视化方法之一。它能够将高维的基因表达数据投影到二维空间,保留细胞间的相似性关系。
可视化内容:
- 按样本分组(
SAMple):展示不同样本的细胞在降维空间中的分布,用于评估批次校正效果。 - 按聚类分组(
Seurat_clusters):展示不同细胞群体的分布,用于识别细胞类型和亚群。 - 细胞类型分组(
celltype):通过组织细胞类型特异性 marker 进行区分。
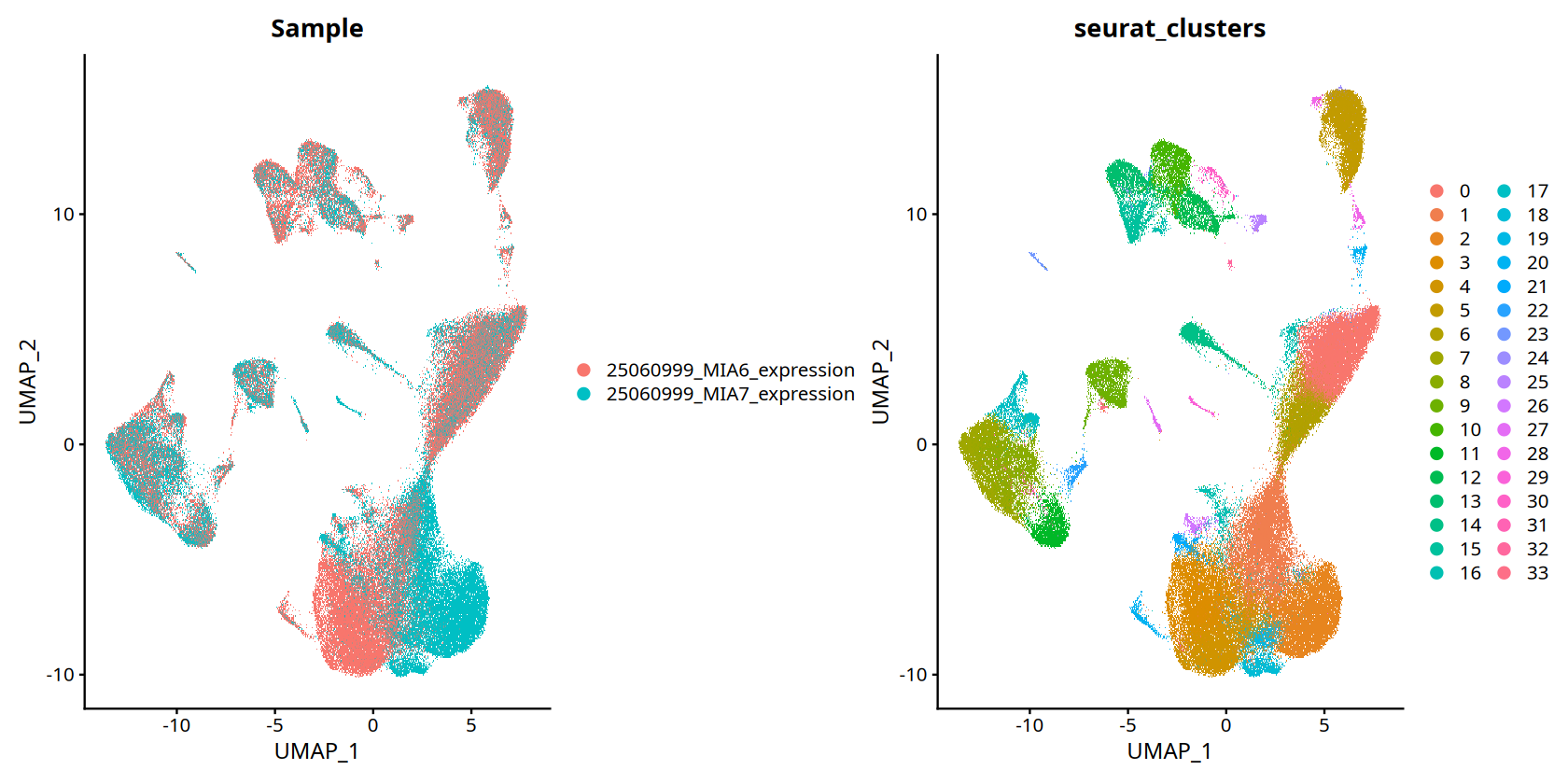
图注说明(UMAP 降维可视化图):
下图展示了细胞在降维空间中的分布情况,按样本或聚类分组显示。
- 点的含义:每个点代表一个细胞。
- 距离关系:距离相近的点表示细胞表达谱相似。
- 颜色分组:相同颜色或标记的点属于同一类别(样本或细胞类型)。
- 批次校正评估:不同样本的相同细胞类型应该聚集在一起,说明批次校正成功。
# --- UMAP 降维可视化 ---
# options(): 设置图形输出的尺寸(高度和宽度,单位为英寸)
# repr.plot.height 和 repr.plot.width: 控制 Jupyter notebook 中图形显示的大小
options(repr.plot.height=7, repr.plot.width=14)
# DimPlot(): Seurat 中的降维可视化函数
# 参数说明:
# - obj.integrated: Seurat 对象(如果您的变量名是 data,请替换为 data)
# - reduction = 'umap': 使用 UMAP 降维结果进行可视化
# - group.by: 指定分组变量,可以是向量(c())包含多个分组方式
# * "stim": 按样本分组显示
# * "RNA_snn_res.0.8": 按聚类结果分组显示(分辨率 0.8)
DimPlot(data, reduction = 'umap', group.by=c("Sample","seurat_clusters"))To disable this behavior set \`raster=FALSE\`
Rasterizing points since number of points exceeds 100,000.
To disable this behavior set \`raster=FALSE\`
空间位置可视化
功能概述:
空间转录组数据的核心优势在于能够保留细胞的原始空间位置信息。通过将 reduction 参数设置为 "spatial",我们可以将细胞绘制在组织样本的实际空间坐标上,观察不同细胞类型或特征在组织中的分布模式。
可视化内容:
- 展示细胞在组织切片中的实际空间位置。
- 可以按样本、聚类结果或其他元数据分组显示。
- 用于分析细胞类型的空间分布规律,识别空间异质性。
参数说明:
reduction = 'spatial':使用空间坐标进行可视化。subset = stim=="样本名":筛选特定样本进行可视化。pt.size:控制点的尺寸,根据细胞密度调整。
应用场景:
- 识别细胞类型的空间分布模式。
- 分析不同区域的组织结构。
- 研究空间相关的生物学过程。
# --- 样本 MIA6 的空间位置可视化 ---
# 设置图形输出尺寸
options(repr.plot.height=7, repr.plot.width=15)
# subset(): 筛选特定样本的数据
# - obj.integrated: Seurat 对象
# - subset = stim=="MIA6": 筛选样本标识为 "MIA6" 的细胞
# DimPlot(): 空间可视化
# - reduction = 'spatial': 使用空间坐标进行可视化
# - pt.size = 1: 设置点的尺寸(可根据需要调整,如 0.5-2.0)
DimPlot(subset(data,subset=Sample=="25060999_MIA6_expression"), reduction = 'spatial',pt.size = 1)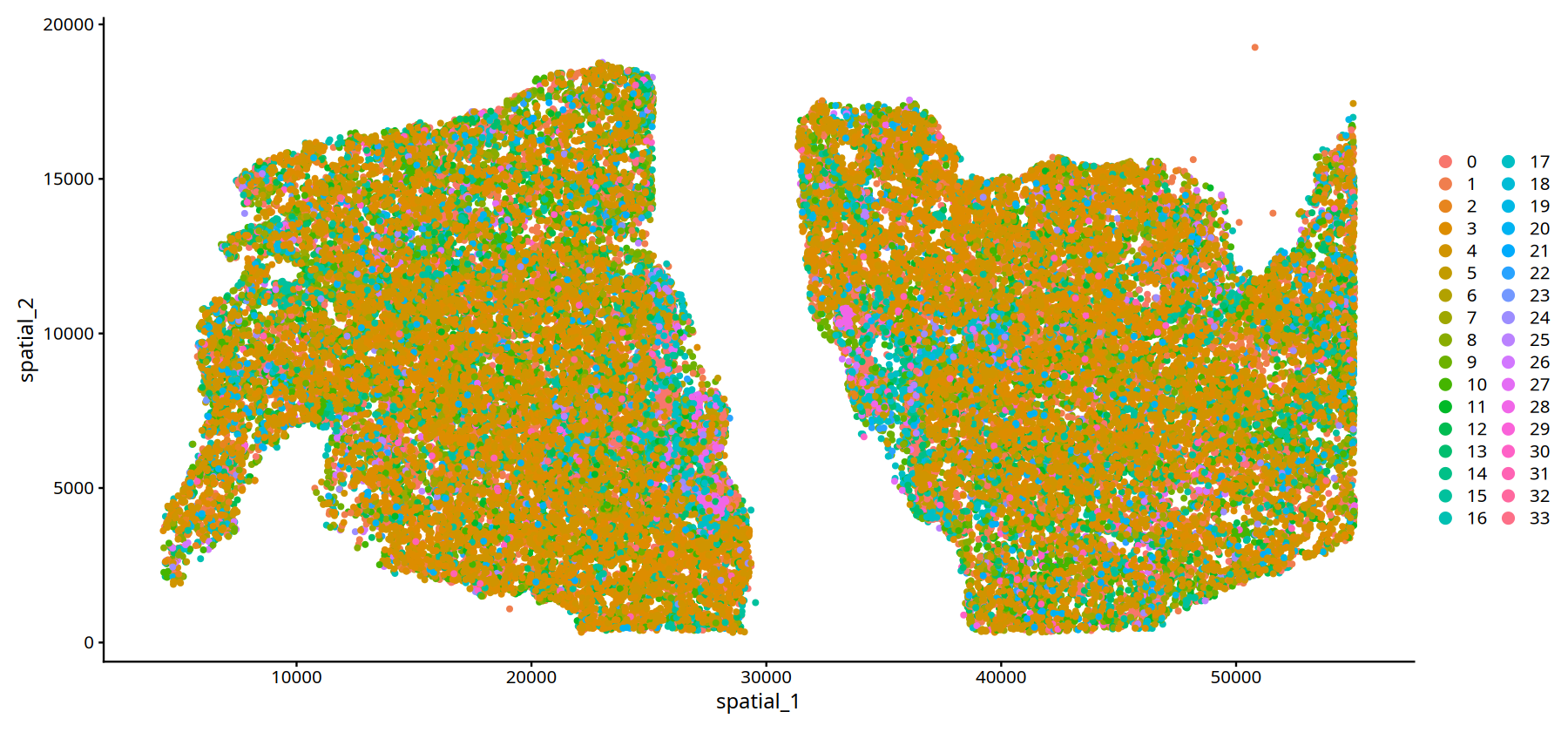
# --- 样本 MIA7 的空间位置可视化 ---
# 设置图形输出尺寸
options(repr.plot.height=7, repr.plot.width=15)
# 筛选样本标识为 "MIA7" 的细胞,并绘制空间位置图
# 参数说明同上述代码
DimPlot(subset(data,subset=Sample=="25060999_MIA7_expression"), reduction = 'spatial',pt.size = 1)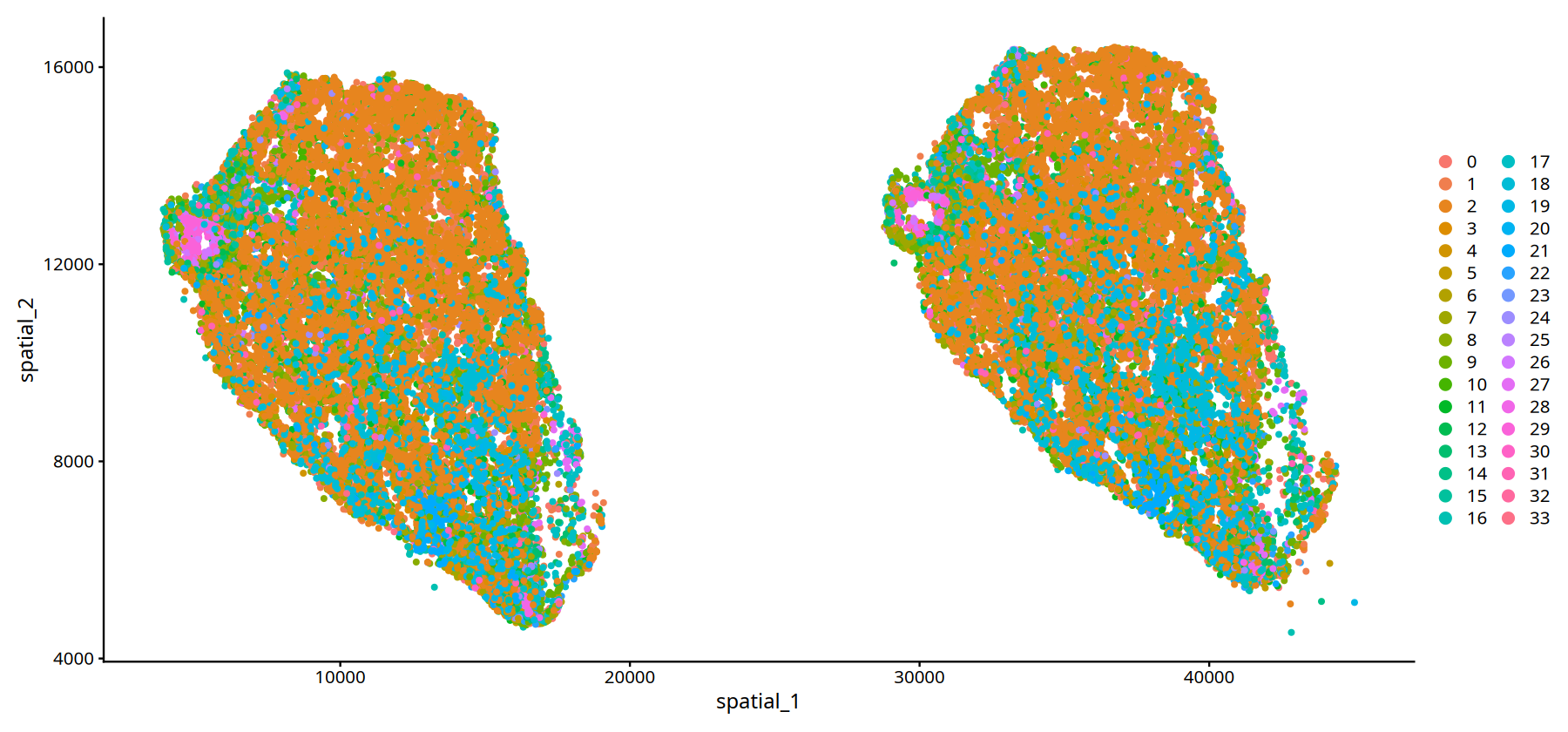
自定义空间绘图函数
为了更灵活地展示空间转录组数据,我们自定义了两个绘图函数,可以在组织图像背景上叠加细胞信息:
函数功能:
ImageSpacePlot:绘制细胞分群信息- 在组织图像(HE 染色或 DAPI 染色)背景上展示细胞的聚类结果。
- 不同颜色代表不同的细胞类型/聚类。
- 适用于展示细胞类型的空间分布模式。
FeatureSpacePlot:绘制连续变量(如基因表达)- 在组织图像背景上展示连续变量的分布(如基因表达量)。
- 使用颜色梯度表示数值大小。
- 适用于展示特定基因或特征的空间表达模式。
使用优势:
- 结合组织形态学信息,更直观地理解数据。
- 可以选择不同的背景图像类型(HE 或 DAPI)。
- 可以灵活调整点的大小、透明度和颜色方案。
# ============================================================================
# 自定义空间绘图函数定义
# ============================================================================
# --- ImageSpacePlot 函数:绘制细胞分群信息 ---
# 功能:在组织图像背景上绘制细胞的聚类/分群结果
#
# 参数说明:
# - obj: Seurat 对象,包含空间转录组数据
# - group_by: 字符串,指定用于分组的元数据列名(如聚类结果列名)
# - type: 字符串,"DAPI" 或 "HE",指定背景图像类型
# - sample: 字符串,指定要可视化的样本名称(默认使用第一个样本)
# - size: 数值,控制点的尺寸(默认 1)
# - alpha: 数值,控制点的透明度(0-1,默认 1 为完全不透明)
# - color: 颜色向量,自定义颜色方案(默认使用预定义的 MYCOLOR)
#
ImageSpacePlot = function(obj, group_by, type="DAPI", sample=names(obj@misc$info)[1], size=1, alpha=1,color=MYCOLOR){
# 定义默认颜色方案(40种颜色,足够大多数聚类结果使用)
MYCOLOR=c(
"#6394ce", "#2a4c87", "#eed500", "#ed5858",
"#f6cbc2", "#f5a2a2", "#3ca676", "#6cc9d8",
"#ef4db0", "#992269", "#bcb34a", "#74acf3",
"#3e275b", "#fbec7e", "#ec4d3d", "#ee807e",
"#f7bdb5", "#dbdde6", "#f591e1", "#51678c",
"#2fbcd3", "#80cfc3", "#fbefd1", "#edb8b5",
"#5678a8", "#2fb290", "#a6b5cd", "#90d1c1",
"#a4e0ea", "#837fd3", "#5dce8b", "#c5cdd9",
"#f9e2d6", "#c64ea4", "#b2dfd6", "#dbdfe7",
"#dff2ec", "#cce8f3", "#e74d51", "#f7c9c4",
"#f29c81", "#c9e6e0", "#c1c5de", "#750000"
)
# 根据图像类型选择对应的栅格图像数据
raster_type <- switch(type,
HE = "img_he_gg", # HE 染色图像
DAPI = "img_gg", # DAPI 染色图像
stop("Invalid type. Must be 'HE' or 'DAPI'.")
)
# 提取分组信息(如聚类结果)
spatial_coord1 <- as.data.frame(obj[[group_by]])
colnames(spatial_coord1) <- group_by
# 提取空间坐标信息
spatial_coord2 <- as.data.frame(obj@reductions$spatial@cell.embeddings)
# 合并空间坐标和分组信息
spatial_coord <-cbind(spatial_coord2,spatial_coord1)
# 使用 ggplot2 绘制图形
# 1. 添加背景图像(annotation_custom)
# 2. 添加细胞点(geom_point),按分组信息着色
ImageSpacePlot <- ggplot2::ggplot() +
ggplot2::annotation_custom(grob = obj@misc$info[[sample]][[raster_type]],
xmin = 0, xmax = obj@misc$info[[sample]]$size_x,
ymin = 0, ymax = obj@misc$info[[sample]]$size_y) +
ggplot2::geom_point(data = spatial_coord,
ggplot2::aes(x = spatial_1, y = spatial_2,
color = !!sym(group_by),
fill = !!sym(group_by)),
size=size, alpha=alpha)+
labs(size = group_by) + guides(alpha = "none")+
ggplot2::theme_classic()+
scale_color_manual(values = color)+ coord_fixed()
return(ImageSpacePlot)
}
# --- FeatureSpacePlot 函数:绘制连续变量(如基因表达)---
# 功能:在组织图像背景上绘制连续变量的空间分布(如基因表达量)
#
# 参数说明:
# - obj: Seurat 对象,包含空间转录组数据
# - feature: 字符串,指定要可视化的特征(如基因名或元数据列名)
# - type: 字符串,"DAPI" 或 "HE",指定背景图像类型
# - sample: 字符串,指定要可视化的样本名称
# - size: 数值,控制点的尺寸
# - alpha: 数值向量,控制透明度的范围(c(最小值, 最大值))
# - color: 颜色向量,定义颜色梯度(c(低值颜色, 高值颜色))
#
FeatureSpacePlot = function(obj, feature, type="DAPI", sample=names(obj@misc$info)[1], size=1, alpha=c(1,1),color=c("lightgrey","blue")){
# 根据图像类型选择对应的栅格图像数据
raster_type <- switch(type,
HE = "img_he_gg",
DAPI = "img_gg",
stop("Invalid type. Must be 'HE' or 'DAPI'.")
)
# 提取空间坐标信息
spatial_coord1 <- as.data.frame(obj@reductions$spatial@cell.embeddings)
# 提取特征数据(如基因表达量)
spatial_coord2 <- FetchData(obj,feature)
colnames(spatial_coord2) <- feature
# 合并空间坐标和特征数据
spatial_coord <-cbind(spatial_coord1,spatial_coord2)
# 使用 ggplot2 绘制图形
# 1. 添加背景图像
# 2. 添加细胞点,按特征值着色和调整透明度
FeatureSpacePlot <-ggplot2::ggplot() +
ggplot2::annotation_custom(grob = obj@misc$info[[sample]][[raster_type]],
xmin = 0, xmax = obj@misc$info[[sample]]$size_x,
ymin = 0, ymax = obj@misc$info[[sample]]$size_y) +
ggplot2::geom_point(data = spatial_coord,
ggplot2::aes(x = spatial_1, y = spatial_2,
color = !!sym(feature),
alpha = !!sym(feature)),
size=size)+
labs(color = feature)+
guides(alpha = "none")+
ggplot2::theme_classic()+
ggplot2::scale_alpha_continuous(range=alpha)+
scale_color_gradient(low=color[1],high = color[2])+ coord_fixed()
return(FeatureSpacePlot)
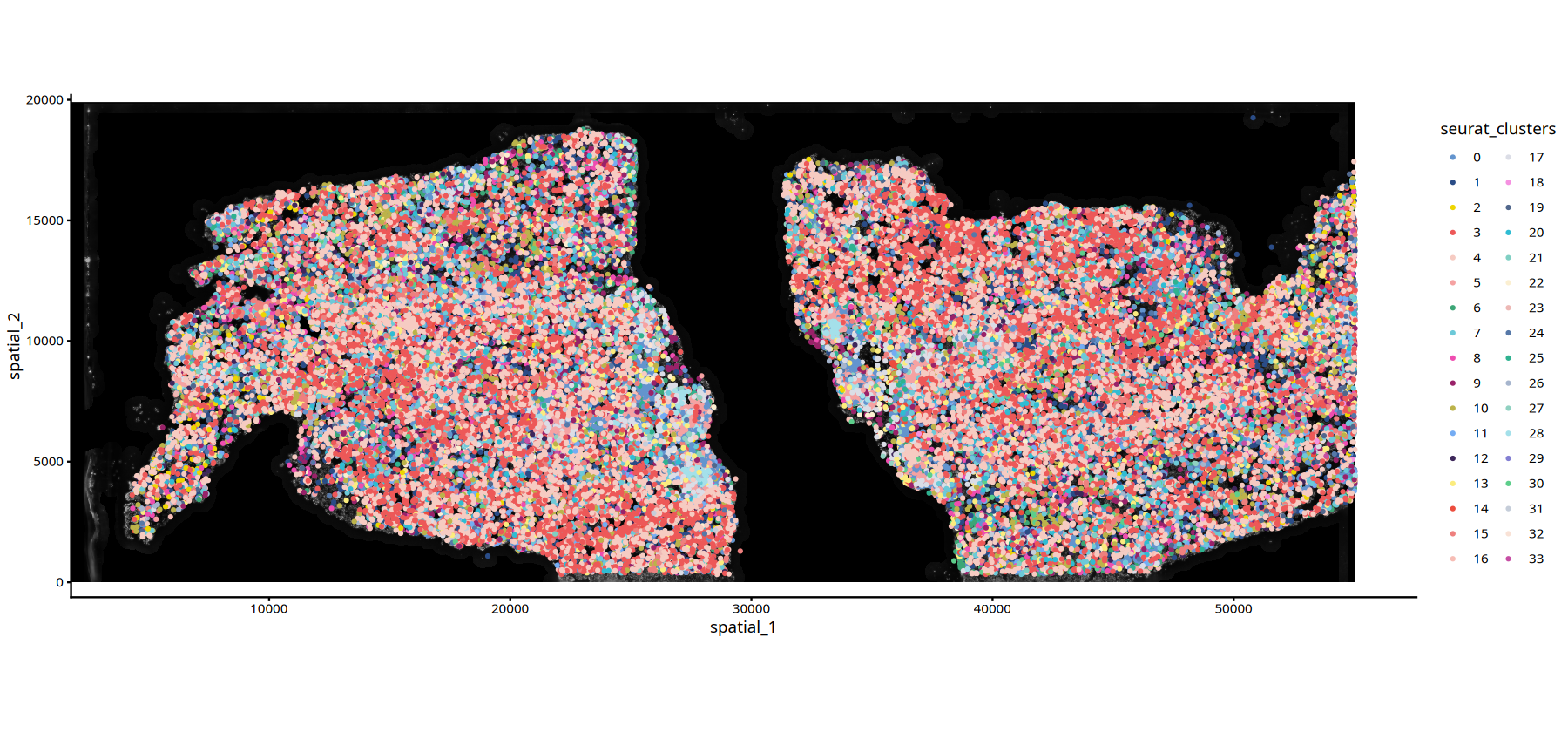
}# --- 使用 ImageSpacePlot 绘制样本 MIA6 的细胞分群图 ---
# 设置图形输出尺寸
options(repr.plot.height=7, repr.plot.width=15)
# ImageSpacePlot(): 自定义函数,在组织图像背景上绘制细胞分群
# 参数说明:
# - subset(): 筛选样本标识为 "MIA6" 的细胞
# - group_by = "seurat_clusters": 按聚类结果分组着色
# - type = "DAPI": 使用 DAPI 染色图像作为背景
# - size = 0.7: 设置点的尺寸为 0.7
# - sample = "25060999_MIA6_expression": 指定样本名称
ImageSpacePlot(subset(data,subset=Sample=="25060999_MIA6_expression"),
group_by = "seurat_clusters",
type="DAPI",
size=0.7,
sample="25060999_MIA6_expression")• size : "seurat_clusters"
# --- 使用 ImageSpacePlot 绘制样本 MIA7 的细胞分群图 ---
# 设置图形输出尺寸
options(repr.plot.height=7, repr.plot.width=15)
# 绘制样本 MIA7 的细胞分群图
# 参数说明同上述代码
ImageSpacePlot(subset(data,subset=Sample=="25060999_MIA7_expression"),
group_by = "seurat_clusters",
type="DAPI",
size=0.7,
sample="25060999_MIA7_expression")• size : "seurat_clusters"

# DAPI背景的基因表达图
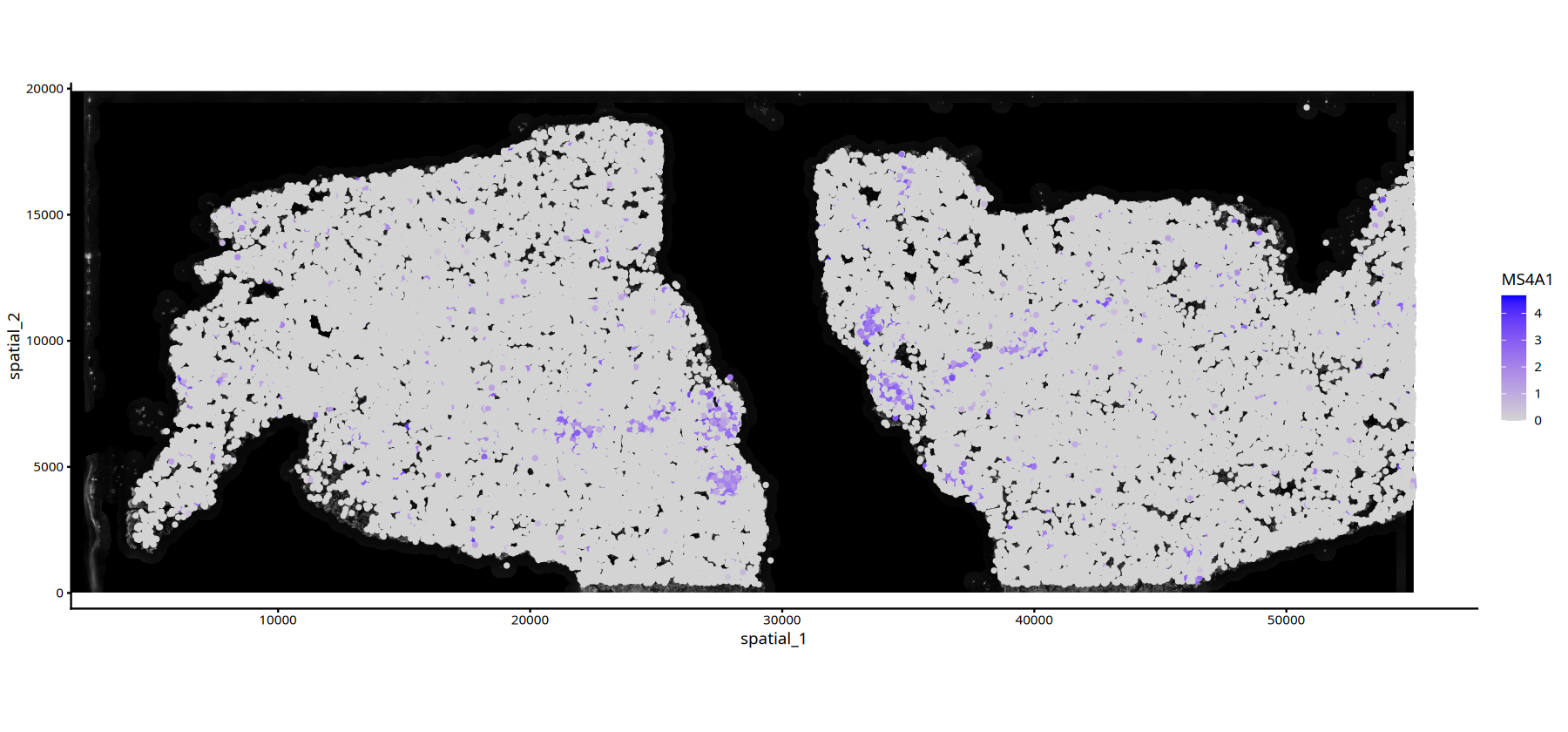
options(repr.plot.height=7, repr.plot.width=15)
FeatureSpacePlot(obj=subset(data,subset=Sample=="25060999_MIA6_expression"), feature="MS4A1",type="DAPI")