ATAC + RNA 多组学:CopyscAT 基于 scATAC 的 CNV 推断与肿瘤细胞识别
文档概览
CopyscAT 可基于 scATAC-seq 数据推断拷贝数变异(CNV),辅助识别癌症细胞。它能帮助研究复杂肿瘤(如胶质母细胞瘤)内不同亚克隆的染色体变化与表观基因组状态的关系,分析遗传变异如何影响细胞的分子表型。CopyscAT 特别适合探索高异质性肿瘤中遗传与表观遗传的交互,以及肿瘤细胞与微环境间的作用机制。
单细胞多组学数据中包含 scATAC-seq 信息,可以利用 CopyscAT 对多组学中的 scATAC-seq 做 CNV 分析,揭示肿瘤细胞异质性。
CNV 分析的意义
什么是 CNV
**拷贝数变异(Copy Number Variation,CNV)**是指基因组中较大 DNA 片段在数量上的结构变异,主要表现为染色体区域的扩增(gain)或缺失(loss)。正常情况下,人类细胞为二倍体(每条常染色体通常有 2 个拷贝);当发生扩增时拷贝数超过 2,发生缺失时拷贝数低于 2。
CNV 是肿瘤等多种疾病发生和进展的重要驱动力。传统的 CNV 分析主要基于全基因组测序(WGS)或全外显子测序(WES),只能提供群体平均水平的信息。单细胞 CNV 分析则在单细胞分辨率下推断每个细胞的拷贝数状态,能够揭示组织内部不同细胞的基因组差异与异质性。
单细胞多组学 CNV 分析的两个方向
单细胞多组学数据用于 CNV 分析主要有两个方向:
- 方向一:基于 scRNA-seq 数据 - 利用基因表达信息推断 CNV,主要工具为 InferCNV
- 方向二:基于 scATAC-seq 数据 - 利用读段数量推断 CNV,主要工具包括 epiAneuFinder、AtaCNV、CopyscAT
TIP
主流单细胞 ATAC-seq CNV 分析工具简介
目前,常用于 scATAC-seq 数据 CNV 检测的工具有:epiAneuFinder、CopyscAT 和 AtaCNV。本指南聚焦 CopyscAT 的详细用法。如需使用其他工具,可参考 AtaCNV 与 epiAneuFinder 的相关文档获取更多信息。
适用场景与主要目的
适合的样本类型:
- 肿瘤组织样本(强烈推荐) - 包含大量 CNV 事件,可揭示肿瘤异质性和亚克隆结构
- 癌前病变或发育异常样本 - 可检测早期基因组结构变异
不适合的样本类型:
- 正常健康组织 - 大多数细胞为二倍体,缺乏显著的 CNV 事件,CNV 分析意义有限
单细胞 CNV 分析的主要目的:
- 恶性细胞识别 - 区分恶性细胞与非恶性细胞(恶性细胞通常表现出大范围的、非随机的 CNV 模式)
- 肿瘤异质性解析 - 识别具有不同 CNV 特征的亚克隆群体
- 克隆进化追踪 - 通过 CNV 模式相似性推断肿瘤的克隆进化关系
CopyscAT 分析的实现
CopyscAT 工具的原理
CopyscAT 使用单细胞表观基因组数据来推断拷贝数变异(CNV),从而定义和识别癌症细胞。该工具的核心原理是利用 scATAC-seq 数据中比对到基因组区域的读段数量作为该区域 DNA 拷贝数的代理指标。
CopyscAT 的分析流程包括以下关键步骤:
- 数据输入:接受由 SeekArc 生成的 barcode-fragment 矩阵作为输入
- 数据标准化:对覆盖度矩阵进行标准化处理,生成标准化矩阵
- CNV 检测:
- 局部 CNV 检测:通过标准化覆盖度矩阵中的大峰值推断局部 CNV(如 ecDNA,即染色体外 DNA)
- 片段和染色体臂水平 CNV 检测:使用标准化矩阵推断片段水平和染色体臂水平的 CNV
- 双分钟(double minutes)检测:识别基因组中的高拷贝数扩增区域
- 细胞分配:使用共识聚类(consensus clustering)方法最终确定细胞的分组和 CNV 状态
CopyscAT 的一个关键优势是能够自动识别非肿瘤细胞并使用它们作为对照,从而更准确地检测肿瘤细胞中的 CNV。该工具特别适合处理具有高水平肿瘤内异质性的复杂样本。
下图为 CopyscAT 主要分析流程示意:
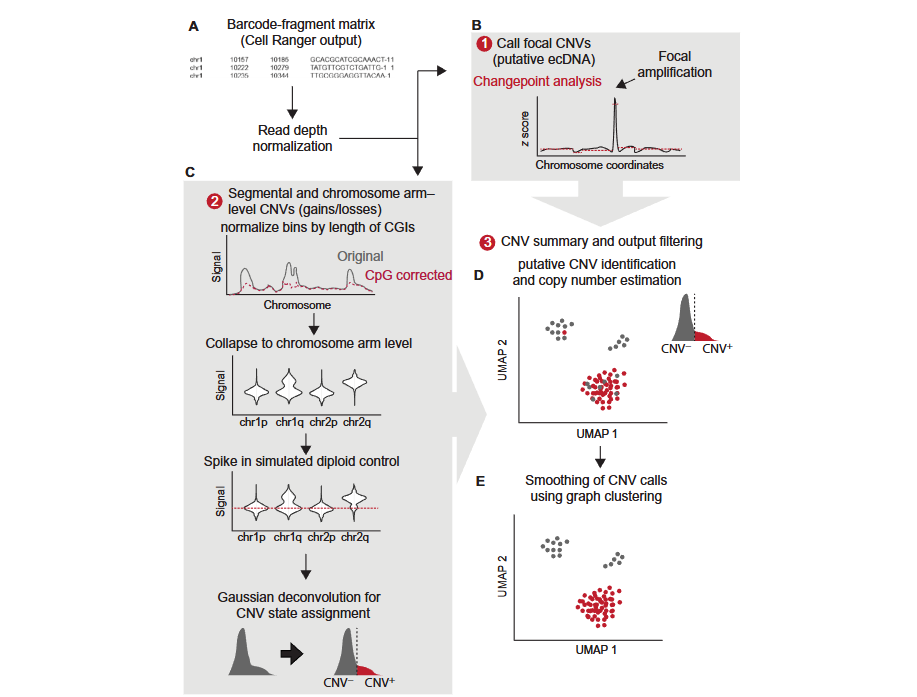
分析步骤
CopyscAT 的分析流程包括初始化设置、数据标准化、CNV 检测和结果输出等步骤。以下是完整的使用示例:
初始一次性设置
library("devtools")
# STEP 1: 替换 "~/CopyscAT" 为您 git clone 的仓库路径
install("~/CopyscAT")
# 或者使用 GitHub 安装(某些 R 版本可能会因代码警告报错,可使用下面这行代码解决)
Sys.setenv("R_REMOTES_NO_ERRORS_FROM_WARNINGS" = "true")
install_github("spcdot/copyscat")
# 加载包
library(CopyscAT)
# 加载您使用的基因组
library(BSgenome.Hsapiens.UCSC.hg38)
# 生成参考文件 - 这将创建基因组 chrom.sizes 文件、cytobands 文件和 CpG 文件
generateReferences(BSgenome.Hsapiens.UCSC.hg38,
genomeText = "hg38",
tileWidth = 1e6,
outputDir = "~")常规工作流程
# 初始化环境(注意:每次运行 CopyscAT 的会话都需要执行此步骤)
initialiseEnvironment(genomeFile="~/hg38_chrom_sizes.tsv",
cytobandFile="~/hg38_1e+06_cytoband_densities_granges.tsv",
cpgFile="~/hg38_1e+06_cpg_densities.tsv",
binSize=1e6,
minFrags=1e4,
cellSuffix=c("-1","-2"),
lowerTrim=0.5,
upperTrim=0.8)
# 设置输出默认目录和文件名
setOutputFile("~","samp_dataset")
# ===== PART 1: 初始数据标准化 =====
# 读取输入数据(如果使用自己的文件,替换为以下代码)
# scData <- readInputTable("myInputFile.tsv")
scData <- scDataSamp # 使用包内示例数据
# 标准化矩阵
scData_k_norm <- normalizeMatrixN(scData,
logNorm = FALSE,
maxZero=2000,
imputeZeros = FALSE,
blacklistProp = 0.8,
blacklistCutoff=125,
dividingFactor=1,
upperFilterQuantile = 0.95)
# 折叠到染色体臂水平
summaryFunction <- cutAverage
scData_collapse <- collapseChrom3N(scData_k_norm,
summaryFunction=summaryFunction,
binExpand = 1,
minimumChromValue = 100,
logTrans = FALSE,
tssEnrich = 1,
logBase=2,
minCPG=300,
powVal=0.73)
# 应用额外过滤
scData_collapse <- filterCells(scData_collapse,
minimumSegments = 40,
minDensity = 0.1)
# 显示未缩放的染色体列表
graphCNVDistribution(scData_collapse, outputSuffix = "test_violinsn2")
# 计算中心
median_iqr <- computeCenters(scData_collapse,
summaryFunction=summaryFunction)
# ===== PART 2: 染色体水平 CNV 评估 =====
# OPTION 1: 使用所有细胞生成"正常"对照来识别染色体水平扩增
candidate_cnvs <- identifyCNVClusters(scData_collapse,
median_iqr,
useDummyCells = TRUE,
propDummy=0.25,
minMix=0.01,
deltaMean = 0.03,
deltaBIC2 = 0.25,
bicMinimum = 0.1,
subsetSize=600,
fakeCellSD = 0.08,
uncertaintyCutoff = 0.55,
summaryFunction=summaryFunction,
maxClust = 4,
mergeCutoff = 3,
IQRCutoff= 0.2,
medianQuantileCutoff = 0.4)
# 清理步骤
candidate_cnvs_clean <- clusterCNV(initialResultList = candidate_cnvs,
medianIQR = candidate_cnvs[[3]],
minDiff=1.5)
# 最终结果和注释
final_cnv_list <- annotateCNV4(candidate_cnvs_clean,
saveOutput=TRUE,
outputSuffix = "clean_cnv",
sdCNV = 0.5,
filterResults=TRUE,
filterRange=0.8)
# OPTION 2: 自动识别非肿瘤细胞并使用它们作为对照
# 注意:该方法仍在实验阶段,如果选项 A 的拷贝数调用不合理时使用 (例如基线似乎不正确)
# 如果肿瘤细胞纯度 <90%,效果最佳 (非常小的非肿瘤细胞群体可能会被遗漏)
# 运行此步骤需要在 filterCells 之后
nmf_results <- identifyNonNeoplastic(scData_collapse,
methodHclust="ward.D",
cutHeight = 0.4)
# 保存非肿瘤细胞识别结果
write.table(x=rownames_to_column(data.frame(nmf_results$cellAssigns),
var="Barcode"),
file=str_c(scCNVCaller$locPrefix,
scCNVCaller$outPrefix,
"_nmf_clusters.csv"),
quote=FALSE,
row.names = FALSE,
sep=",")
print(paste("Normal cluster is: ",nmf_results$clusterNormal))
# 使用正常细胞作为背景的替代 CNV 调用方法
# 仅基于正常细胞计算中心趋势
median_iqr <- computeCenters(scData_collapse %>%
select(chrom,nmf_results$normalBarcodes),
summaryFunction=summaryFunction)
# 使用正常细胞进行 CNV 调用
candidate_cnvs <- identifyCNVClusters(scData_collapse,
median_iqr,
useDummyCells = TRUE,
propDummy=0.25,
minMix=0.01,
deltaMean = 0.03,
deltaBIC2 = 0.25,
bicMinimum = 0.1,
subsetSize=800,
fakeCellSD = 0.09,
uncertaintyCutoff = 0.65,
summaryFunction=summaryFunction,
maxClust = 4,
mergeCutoff = 3,
IQRCutoff = 0.25,
medianQuantileCutoff = -1,
normalCells=nmf_results$normalBarcodes)
candidate_cnvs_clean <- clusterCNV(initialResultList = candidate_cnvs,
medianIQR = candidate_cnvs[[3]],
minDiff=1.0)
final_cnv_list <- annotateCNV4(candidate_cnvs_clean,
saveOutput=TRUE,
outputSuffix = "clean_cnv",
sdCNV = 0.6,
filterResults=TRUE,
filterRange=0.4)
# 或者使用 annotateCNV4B 并输入 normalBarcodes
final_cnv_list <- annotateCNV4B(candidate_cnvs_clean,
nmf_results$normalBarcodes,
saveOutput=TRUE,
outputSuffix = "clean_cnv_b2",
sdCNV = 0.6,
filterResults=TRUE,
filterRange=0.4,
minAlteredCellProp = 0.5)
# ===== PART 2B: 使用聚类平滑 CNV 调用 =====
smoothedCNVList <- smoothClusters(scDataSampClusters,
inputCNVList = final_cnv_list[[3]],
percentPositive = 0.4,
removeEmpty = FALSE)
# ===== PART 3: 识别双分钟/扩增 =====
# 注意:此步骤较慢,可能需要约 5 分钟
library(compiler)
dmRead <- cmpfun(identifyDoubleMinutes)
# minThreshold 是一个节省时间的选项,不会在任何最大 Z 分数小于 4 的细胞上调用断点
dm_candidates <- dmRead(scData_k_norm,
minCells=100,
qualityCutoff2 = 100,
minThreshold = 4)
write.table(x=dm_candidates,
file=str_c(scCNVCaller$locPrefix,
scCNVCaller$outPrefix,
"samp_dm.csv"),
quote=FALSE,
row.names = FALSE,
sep=",")
# ===== PART 4: 调用片段缺失/扩增 (仅在具有 NMF 检测的正常细胞的数据集上) =====
altered_segments <- getAlteredSegments(scData_k_norm,
nmf_results,
lossThreshold = 0.6)
write.table(x=altered_segments,
file=str_c(scCNVCaller$locPrefix,
scCNVCaller$outPrefix,
"samp_segments.csv"),
quote=FALSE,
row.names = FALSE,
sep=",")
# ===== PART 5: 量化循环细胞 =====
# 使用特定染色体的信号 (我们使用 X 染色体,因为在我们的样本中通常不会被改变)
# 如果在您的样本中 X 染色体有已知改变,请尝试使用不同的染色体
barcodeCycling <- estimateCellCycleFraction(scData,
sampName="sample",
cutoff=1000)
write.table(barcodeCycling[order(names(barcodeCycling))]==max(barcodeCycling),
file=str_c(scCNVCaller$locPrefix,
scCNVCaller$outPrefix,
"_cycling_cells.tsv"),
sep="\t",
quote=FALSE,
row.names=TRUE,
col.names=FALSE)TIP
CopyscAT 分析要点
CopyscAT 提供了两种 CNV 检测策略:
- 选项 1 (推荐):使用所有细胞生成对照,适用于大多数情况
- 选项 2:自动识别非肿瘤细胞作为对照,适用于肿瘤纯度较低(<90%)的样本
关键参数说明:
- binSize:基因组窗口大小,默认 1Mb
- minFrags:每个细胞的最小片段数,默认 10000
- blacklistProp:黑名单区域过滤比例,默认 0.8
- cutHeight:NMF 聚类时的切割高度,需要根据实际数据调整
建议先用默认参数运行,然后根据结果质量进行适当调整。
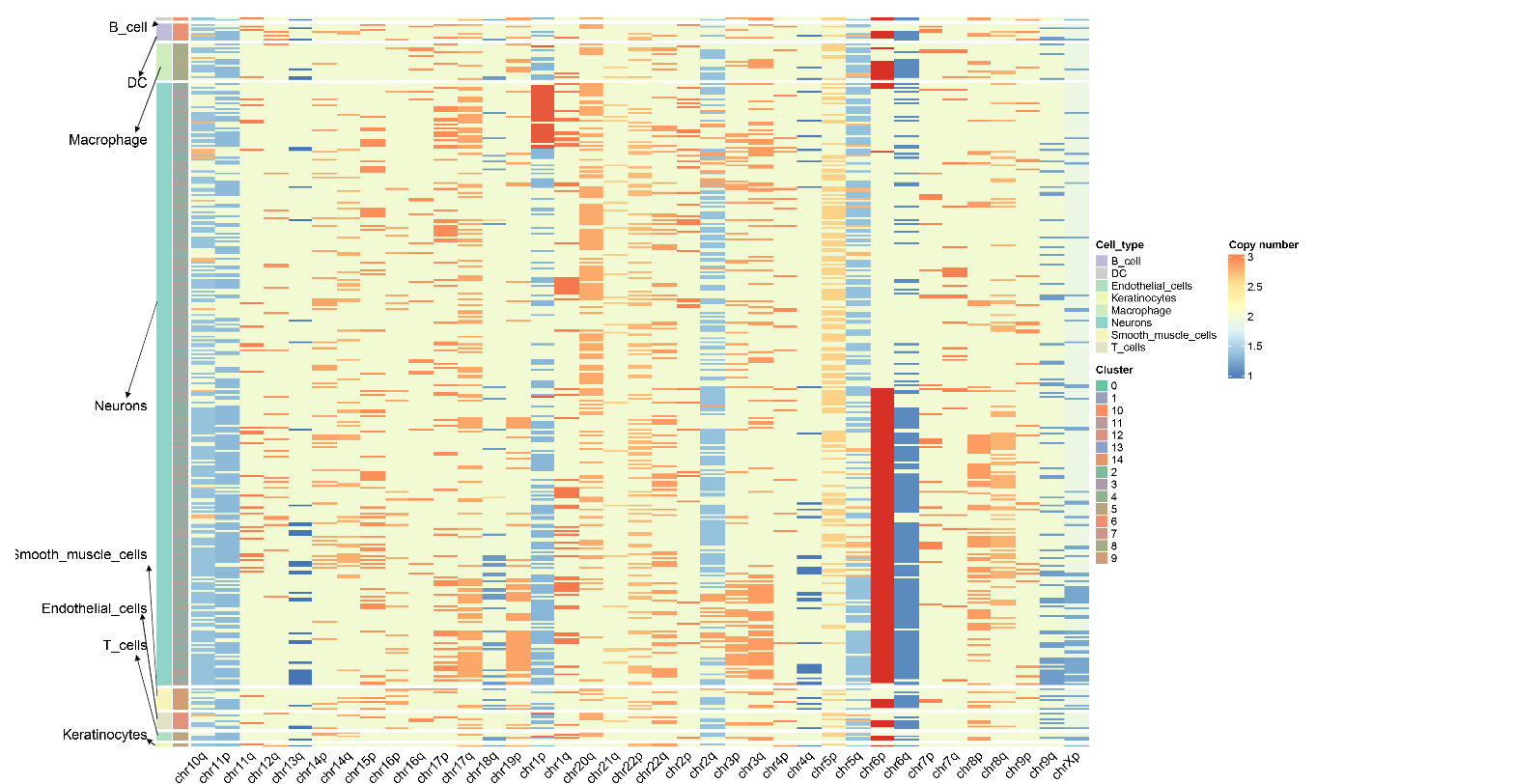
CNV 结果展示--拷贝数变异热图
拷贝数变异热图是 CNV 分析的核心可视化结果,全面展示了所有细胞在全基因组范围内的拷贝数状态:
行与列的含义:
- 每一行代表一个细胞
- 每一列代表一个染色体臂,热图的颜色表示 CNV 分数
- 列的顺序对应基因组从 1 号染色体到性染色体(若保留)的线性排列
颜色映射:
- copy number 值在 2 附近,表示正常的二倍体状态
- copy number 在 1~2 之间,表示染色质存在缺失,越靠近 1,表示缺失越严重
- copy number 在 2~3 之间,表示染色质存在扩增,越靠近 3 表示扩增越严重
- 颜色越红表示扩增越严重,颜色越蓝表示缺失越严重
细胞注释:
热图左侧色带分别表示将细胞按照细胞类型和细胞聚类情况进行排列
CopyscAT 使用共识聚类方法最终确定细胞的 CNV 状态和分组
恶性细胞通常表现出明显的 CNV 模式(大范围的扩增或缺失),而非恶性细胞(如 T 细胞、B 细胞)则保持相对正常的二倍体状态
常见问题
Q1:如何选择合适的 CNV 检测策略?
A:CopyscAT 提供两种 CNV 检测策略:
- 选项 1:使用所有细胞生成对照。适用于大多数情况,特别是当样本中肿瘤细胞占比较高时
- 选项 2:自动识别非肿瘤细胞作为对照。适用于肿瘤纯度较低(<90%)的样本,能够更准确地设置基线。但需要注意的是,如果非肿瘤细胞群体非常小,可能会被遗漏
Q2:如何解释拷贝数结果中的数值?
A:在拷贝数推断结果中:
- 接近 2:表示拷贝数正常(二倍体状态),即该区域在对应细胞中保持正常的拷贝数
- 1~2 之间:表示拷贝数缺失(loss),该区域在对应细胞中的拷贝数低于正常水平,越接近 1 表示缺失越严重
- 2~3 之间:表示拷贝数扩增(gain),该区域在对应细胞中的拷贝数高于正常水平,越接近 3 表示扩增越严重
- 这些数值反映了每个细胞在每个基因组窗口的拷贝数状态,可以用于识别恶性细胞和亚克隆结构
Q3:如何调整 NMF 聚类的参数?
A:当使用 identifyNonNeoplastic 函数时,如果结果不理想,可以调整以下参数:
- cutHeight:切割树状图的高度,默认 0.6。如果聚类分离不好,可以降低该值(如 0.4)
- nmfComponents:NMF 组件数量,默认 5。可以根据样本复杂度调整
- estimatedCellularity:估计的肿瘤细胞比例,默认 0.8。如果已知样本的肿瘤纯度,可以调整此参数
Q4:CopyscAT 与其他 CNV 检测工具相比有什么优势?
A:CopyscAT 的主要优势包括:
- 自动识别正常细胞:能够自动识别样本中的非肿瘤细胞并用作对照,提高 CNV 检测的准确性
- 多层次 CNV 检测:能够检测局部 CNV(如 ecDNA)、片段水平 CNV 和染色体臂水平 CNV
- 双分钟检测:能够识别基因组中的高拷贝数扩增区域(double minutes)
- 细胞周期分析:提供细胞周期分数估计功能,有助于理解细胞状态
- 适合复杂肿瘤:特别适合处理具有高水平肿瘤内异质性的复杂样本,如胶质母细胞瘤
Q5:如何处理大样本数据?
A:对于非常大的数据集:
- 可以对数据进行子集抽样(如使用
subsetSize参数) - 可以在不同细胞群体上分别运行分析,然后合并结果
- 对于双分钟检测等耗时步骤,可以考虑只对特定细胞群体运行
参考资料
[1] NIKOLIC A, SINGHAL D, ELLESTAD K, et al. Copy-scAT: Deconvoluting single-cell chromatin accessibility of genetic subclones in cancer[J]. Science Advances, 2021, 7(42): eabg6045.