甲基化 + RNA 双组学质控报告说明文档
时长: 33 分钟
字数: 9.4k 字
更新: 2026-07-20
阅读: 0 次
本文档详细说明了 SeekMethyl 质控报告中各项指标的定义、计算方法以及解读指南,旨在帮助用户理解测序数据与文库质量状况,并为后续数据分析提供参考依据。
报告主要分为三个部分:Joint (联合分析)、RNA (转录组) 和 MET (甲基化)。
Joint Metrics (联合分析指标)
此部分展示基于 RNA 与甲基化双重质控后,且 Barcode 能够一一对应匹配的细胞群体的关键质量与规模指标。
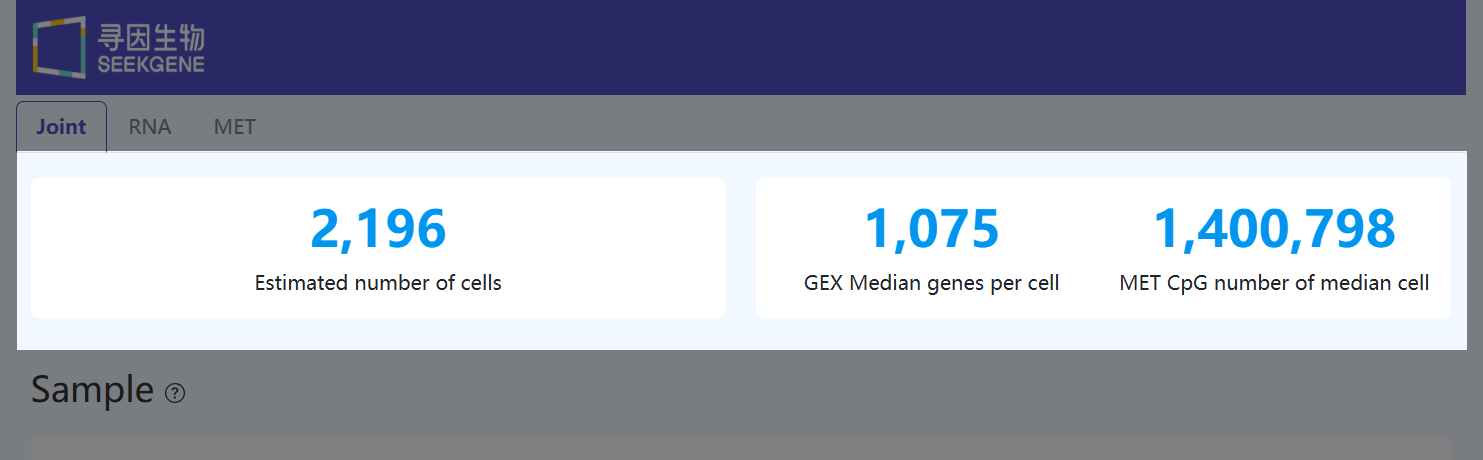
Estimated Number of Cells (有效细胞数,Joint)
- 定义: 同时在 RNA和甲基化数据中均被识别为有效细胞的 Barcode 数量。
- 计算方法: 分别基于 RNA 数据的基因数和 UMI 数,以及甲基化数据的基因组覆盖度和 CpG 数进行细胞识别,并统计在两种组学中均通过质控的 Barcode 交集数量。
- 解读: 该指标表示可用于多组学联合分析的有效细胞规模,是联合分析的基础。
GEX Median Genes per Cell (RNA 中位基因数,Joint)
(该指标与 RNA Cells 模块中的 “Median Genes per Cell” 定义一致)
- 定义: 有效细胞中,每个细胞被检测到的基因数量的中位数。
- 计算方法: 统计所有有效细胞中每个细胞具有至少一个 UMI 计数的基因数,并对该分布取中位数。
- 解读: 该指标反映 RNA 文库的检测灵敏度和复杂度,数值越高通常表示文库质量较好或测序深度较高,从而能够检测到更多低表达基因。
MET CpG Number of Median Cell (甲基化中位细胞 CpG 数,Joint)
(该指标与 MET Cells 模块中的 “CpG Number of Median Cell” 定义一致)
- 定义: 基因组覆盖度处于中位水平的细胞中,被检测到的 CpG 位点数量。
- 计算方法: 根据每个细胞的基因组覆盖度对有效细胞排序,选取覆盖度位于中位的细胞,并统计该细胞中至少被 1 条通过过滤并成功比对的 reads 覆盖、且产生有效甲基化判定的 CpG 位点总数(正负链分别计数)。
- 解读: 该指标反映一个具有代表性的单细胞在甲基化数据中的信息量水平,数值越高表示该细胞在其覆盖范围内检测到的 CpG 位点越多,单细胞层面的甲基化信息相对更丰富。
Barcode Rank Plot for Joint Cell Calling(联合细胞判定的 Barcode–UMI 排序曲线,Joint)
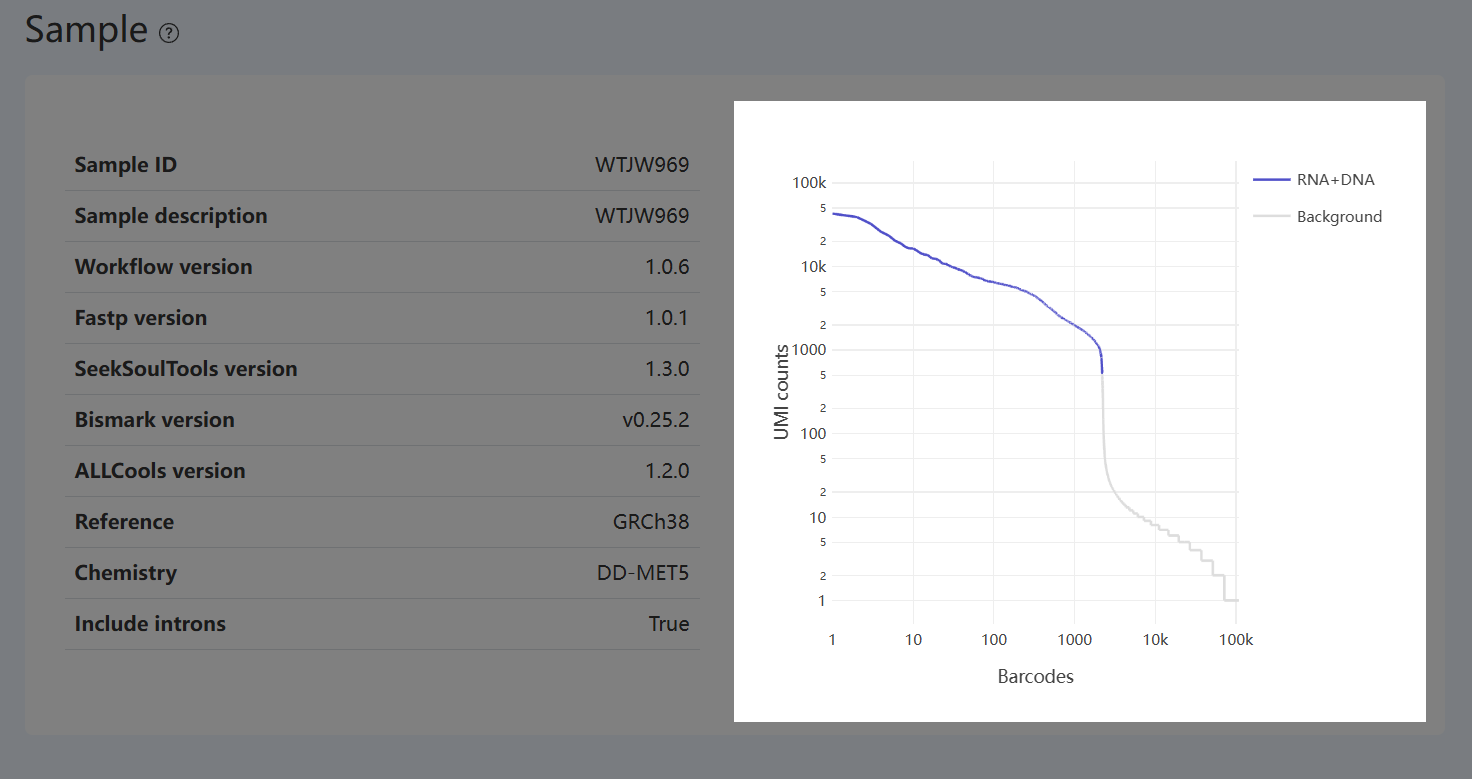
- 定义: 该图展示所有 Barcode 在联合分析流程中用于细胞识别的计数指标分布(以 UMI counts 为例),并按该计数从高到低排序。横轴为排序后的 Barcode(排序位置),纵轴为对应的计数水平。不同颜色用于区分在联合质控与匹配过程中被归类为 RNA+DNA(同时在 RNA 与甲基化数据中均通过细胞识别与质控,且可一一匹配的 Barcode)与 Background 的 Barcode 群体。
- 解读: 该图用于直观查看联合细胞识别过程中,不同类型 Barcode 在计数强度分布上的整体分离情况。
- 分布趋势: 计数强度较高的区间主要由有效细胞相关的 Barcode 构成,而在计数强度较低的区间,Background Barcode 的占比逐渐增加。
- RNA+DNA 曲线: 其在整体分布中的覆盖范围,可用于观察通过联合筛选后、用于多组学联合分析的细胞在全部 Barcode 中所处的位置及规模。
- 整体核查: 不同类型 Barcode 在高、低计数强度区间内的分布关系,以及整体曲线形态,可作为联合细胞识别与筛选结果合理性的直观核查依据。
Feature-based Joint Cell Visualization(基于特征切换的联合细胞可视化,Joint)
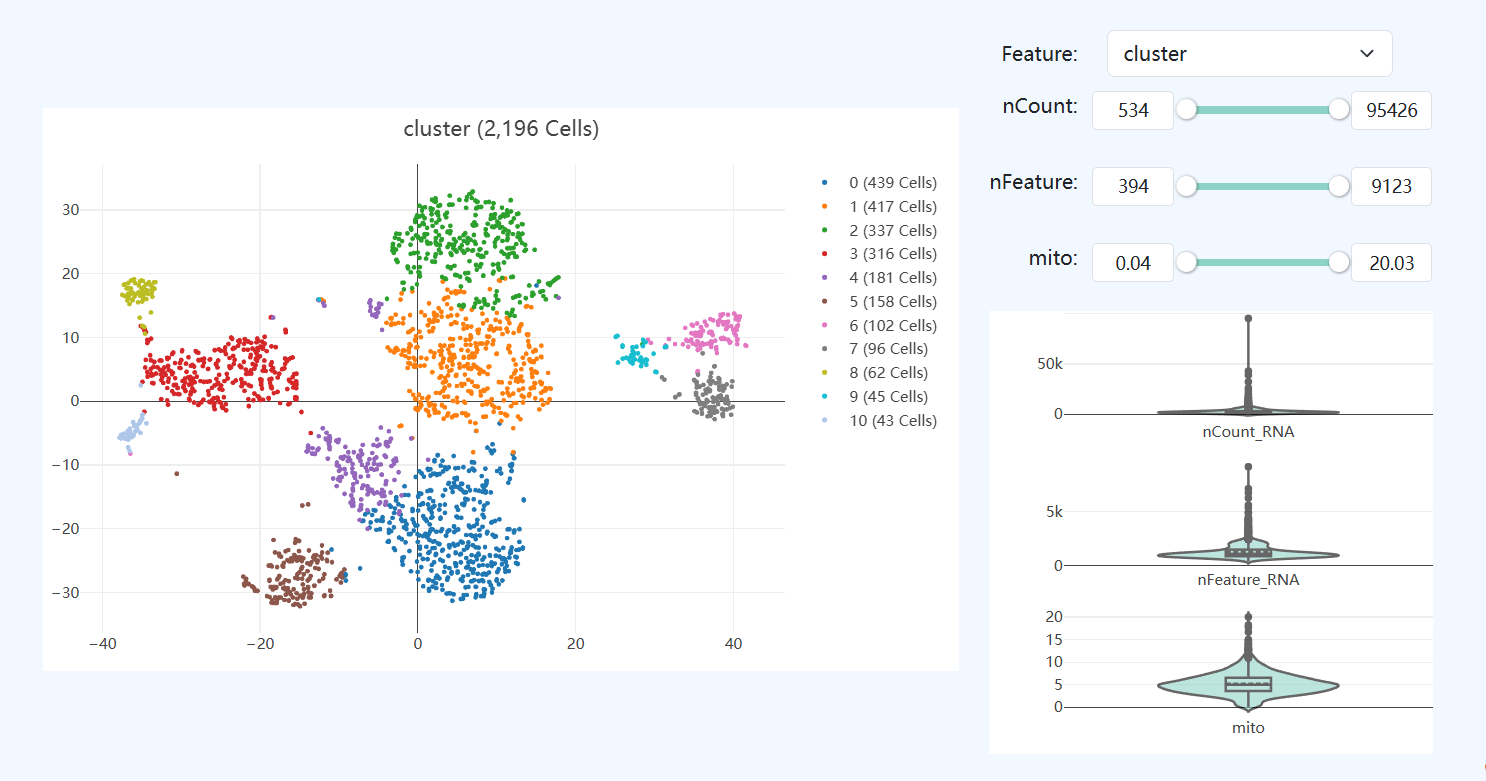
定义: 该模块支持在统一的低维嵌入空间中,根据不同 Feature(特征) 对联合细胞进行着色展示。用户可在 Feature 下拉菜单中选择不同的转录组或甲基化相关指标,以观察对应特征在细胞群体中的分布情况。右侧同时展示所选特征在全体细胞中的分布概览,用于辅助整体判断。
解读: 该可视化用于从多组学角度直观评估联合细胞在不同质量指标和生物学特征下的分布一致性与异质性。
- Cluster: 当选择 cluster 时,可查看联合分析得到的细胞分群结果及各亚群的空间分布;
- RNA Metrics: 当切换至 RNA 相关特征(如
nCount_RNA、nFeature_RNA、mito)时,可评估不同细胞群在转录组测序深度、基因检测能力及线粒体信号方面的差异; - Methylation Metrics: 当切换至甲基化相关特征(如
CpG_number、CpG_methylation、CH_methylation)时,可观察甲基化信息量及甲基化水平在细胞间的分布情况。
通过在同一嵌入空间中切换不同特征,可辅助判断联合细胞在多组学质量与信号层面是否存在系统性偏差,并为后续筛选或分群解释提供参考。
RNA Metrics (转录组指标)
本部分展示单细胞转录组文库的测序质量、比对情况及关键定量指标, 用于评估 RNA 数据的整体质量和可用性,并辅助判断其是否满足下游分析需求。
Sequencing (测序质量,RNA)
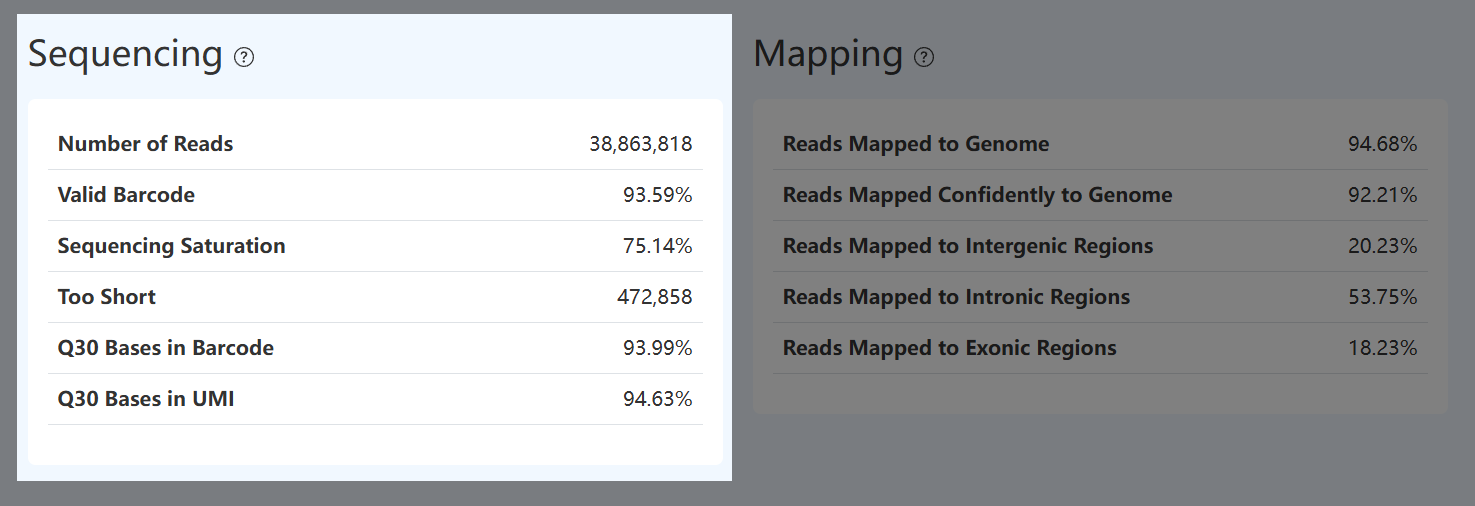
Number of Reads (总 Reads 数,RNA)
- 定义: fastp 质控后保留下来的有效双端 reads pairs 总数。
- 计算方法: 对原始测序数据进行 fastp 质控后,统计通过过滤的 reads pairs 数量。
- 解读: 指标反映样本在质控后可用于后续分析的有效测序数据规模,是下游分析的基础输入。通常需结合有效 Barcode 比例、细胞内 Reads 占比及测序饱和度综合判断,避免仅以测序量评估数据有效性。
Valid Barcode (有效 Barcode 比例,RNA)
- 定义: 在 fastp 质控后的 Reads 中,Barcode 序列能够匹配至细胞白名单的 reads pairs 所占比例。
- 计算方法: 统计 Barcode 序列在无需纠错直接匹配或在允许有限碱基纠错后匹配至白名单的 reads pairs 数量,并除以 Number of Reads。
- 解读: 该指标反映 Barcode 合成质量、测序准确性以及白名单匹配情况。当该指标偏低时,建议结合 Barcode Q30 值及细胞内 Reads 占比一并分析,以区分测序质量问题与文库结构或白名单不匹配的影响。
Sequencing Saturation (测序饱和度,RNA)
- 定义: 用于评估当前测序深度下,文库中分子被重复测序的程度。
- 计算方法: 基于用于 UMI 计数的 reads 计算,定义为 Sequencing Saturation = 1 −(唯一 UMI 数 / 可用于 UMI 计数的 reads 数)。
- 解读: 该指标用于反映测序深度与文库复杂度之间的关系,通常需结合单细胞层面的 UMI 数和基因数共同解读,以评估当前测序深度对信息获取的有效性。一般而言,饱和度升高表示新增测序 reads 中重复分子的比例增加,进一步加深测序对新增分子信息的提升有限;而饱和度较低则说明文库中仍存在较多未被充分采样的分子,增加测序深度仍有助于获得更多新的转录信息。
Too Short (过短 Reads 数量,RNA)
- 定义: 在接头(adapter)去除后,由于 Reads 长度低于最小保留阈值而被过滤掉的 Reads 数量。
- 计算方法: 对 RNA 测序数据进行接头去除后,统计因序列长度低于预设最小长度阈值而被判定为过短并过滤的 Reads 数量。
- 解读: 该指标反映 RNA 文库中因序列过短而被过滤的 Reads 数量情况;当该数量偏高时,通常提示接头残留较多、插入片段偏短或文库构建过程中片段降解,从而导致有效 Reads 在去接头后无法保留,可能进一步影响后续比对效率和定量分析结果。
Q30 Bases in Barcode (Barcode Q30 碱基比例,RNA)
- 定义: Barcode 序列中,碱基测序质量值(Phred score)达到 Q30 及以上的碱基所占比例。
- 计算方法: 统计 Barcode 序列中 Phred 质量值 ≥ 30 的碱基数量,并除以 Barcode 序列的总碱基数量。
- 解读: 该指标反映 Barcode 序列的测序质量水平。较高的 Q30 比例通常表示 Barcode 读取准确性较高,有助于提高有效 Barcode 识别率;当该指标偏低时,可能影响有效细胞的识别数量。
Q30 Bases in UMI (UMI Q30 碱基比例,RNA)
- 定义: UMI 序列中,碱基测序质量值(Phred score)达到 Q30 及以上的碱基所占比例。
- 计算方法: 统计 UMI 序列中 Phred 质量值 ≥ 30 的碱基数量,并除以 UMI 序列的总碱基数量。
- 解读: 该指标反映 UMI 序列的测序质量水平。较高的 Q30 比例通常表示 UMI 识别更为准确,有助于提高分子去重的准确性,从而提升转录本定量结果的可靠性。
Mapping (比对情况,RNA)
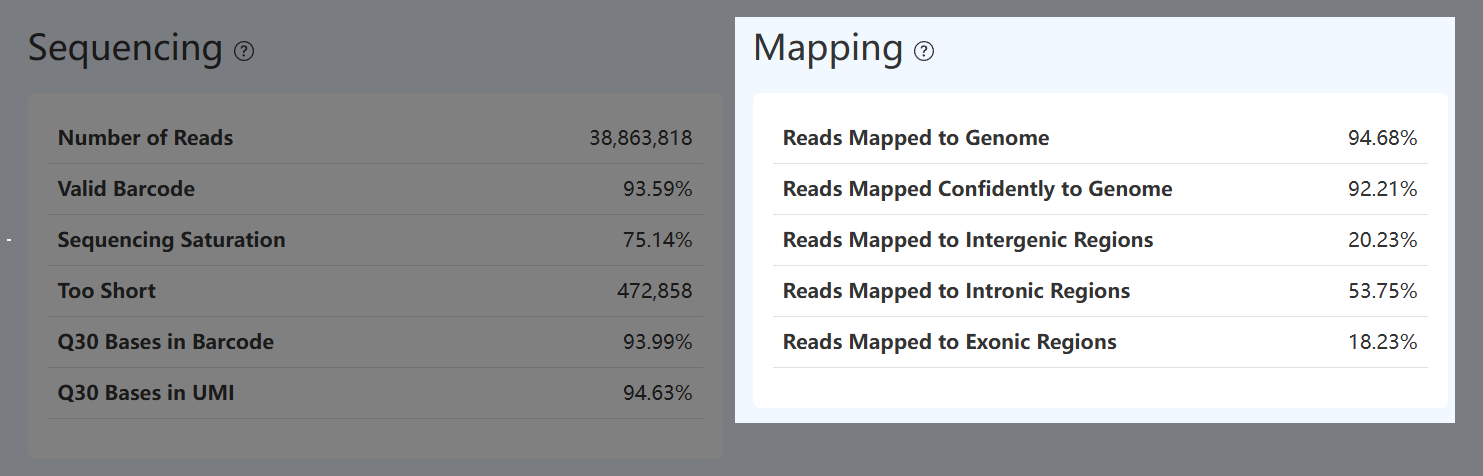
Reads Mapped to Genome (基因组比对率,RNA)
- 定义: 在参与基因组比对的 RNA Reads 中,成功比对到参考基因组的 Reads 所占比例。
- 计算方法: 在完成 Barcode 识别及过短 Reads 过滤后,统计实际参与比对的 RNA Reads 数量;其中成功比对到参考基因组的 Reads 数量(包括唯一比对和多重比对)除以参与比对的 Reads 总数,得到基因组比对率。
- 解读: 该指标反映有效 RNA Reads 与参考基因组的匹配情况及文库特异性。较高的比对率通常表示样本来源明确且参考基因组选择合理;当比对率偏低时,可能与样本污染、低复杂度序列比例偏高或参考基因组版本不匹配有关,通常需结合唯一比对率及外显子/内含子比对分布综合判断比对质量。
Reads Mapped Confidently to Genome (唯一比对率,RNA)
- 定义: 在参与基因组比对的 RNA Reads 中,能够以高置信度唯一比对到参考基因组位置的 Reads 所占比例。
- 计算方法: 在比对结果中筛选 Mapping Quality 较高且仅存在一个最佳比对位置的 Reads,并除以参与比对的 Reads 总数。
- 解读: 该指标反映可用于可靠基因表达定量分析的 Reads 比例。唯一比对率偏低时,可能与重复序列比例较高、测序质量不足或参考基因组不匹配有关,通常需结合总体比对率及外显子比对率共同解读,以评估有效定量信号的占比。
Reads Mapped to Exonic Regions (外显子比对率,RNA)
- 定义: 在成功比对到参考基因组的 RNA Reads 中,比对至已注释外显子区域的 Reads 所占比例。
- 计算方法: 统计所有比对到外显子注释区域的 Reads 数量,并除以成功比对到参考基因组的 Reads 数。
- 解读: 该指标反映转录组文库中成熟 mRNA 信号的富集程度,较高的外显子比对率通常表示文库以成熟转录本为主,有利于基因表达定量分析。
Reads Mapped to Intronic Regions (内含子比对率,RNA)
- 定义: 在成功比对到参考基因组的 RNA Reads 中,比对至已注释内含子区域的 Reads 所占比例。
- 计算方法: 统计所有比对到内含子注释区域的 Reads 数量,并除以成功比对到参考基因组的 Reads 数。
- 解读: 该指标反映未剪接或核内 RNA 信号的占比,其数值受物种、样本类型及建库方式影响,通常需与外显子比对率结合解读,以区分成熟 mRNA 信号与核内转录信号。
Reads Mapped to Intergenic Regions (基因间区比对率,RNA)
- 定义: 在成功比对到参考基因组的 RNA Reads 中,比对至已注释基因间区(intergenic regions)的 Reads 所占比例。
- 计算方法: 统计所有比对到基因间区注释区域的 Reads 数量,并除以成功比对到参考基因组的 Reads 数。
- 解读: 该指标反映 Reads 落在已知基因注释区域之外的比例,数值升高可能与基因组注释不完整、参考注释版本不匹配、非编码转录本或非特异性比对有关。通常需结合总体比对率以及外显子/内含子比对分布等指标综合判断其来源。
Cells (细胞统计,RNA)
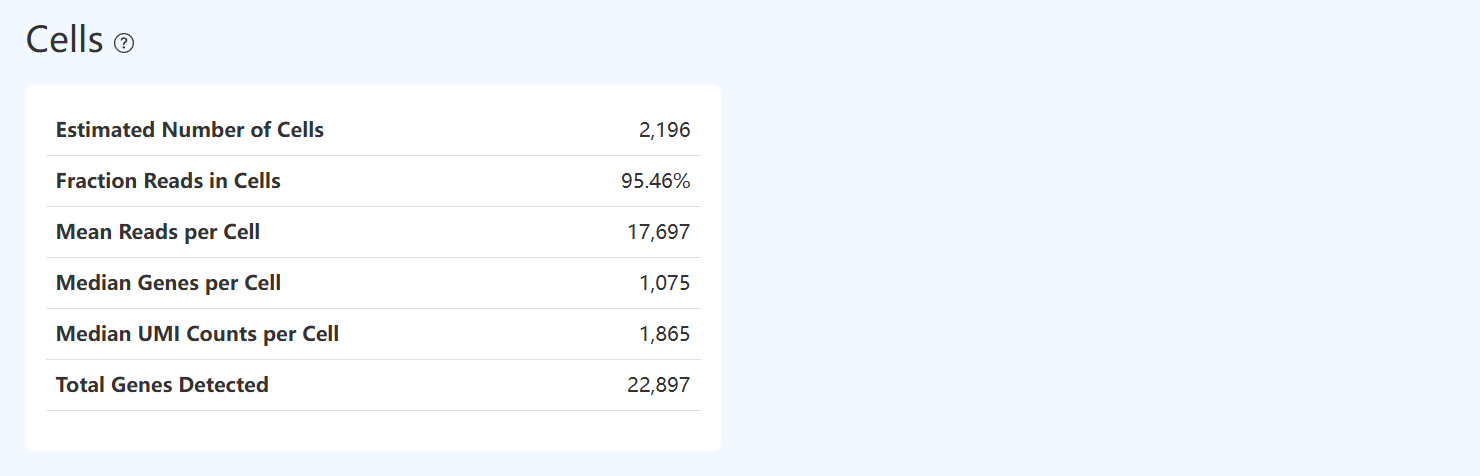
Estimated Number of Cells (有效细胞数,RNA)
- 定义: 基于转录组数据,通过 UMI 去重并结合细胞判定算法识别出的有效细胞 Barcode 数量,即被判定为真实细胞的 Barcode 总数。
- 计算方法: 基于各 Barcode 在 RNA 数据中的 UMI 计数分布,采用细胞判定算法区分真实细胞与背景噪声(如空液滴),统计被识别为有效细胞的 Barcode 数量。
- 解读: 该指标反映转录组测序数据中成功识别的细胞数量。有效细胞数受上样细胞量、测序深度及细胞判定参数等多因素影响,通常需结合平均每个细胞的 Reads 数、细胞内 Reads 占比等 RNA 指标综合评估。
Fraction Reads in Cells (细胞内 Reads 占比,RNA)
- 定义: 在成功比对到参考基因组的 RNA Reads 中,来源于被识别为有效细胞 Barcode 的 Reads 所占比例。
- 计算方法: 在所有成功比对到参考基因组的 Reads 中,统计属于有效细胞 Barcode 的 Reads 数量,并除以成功比对的 Reads 总数。
- 解读: 该指标反映测序数据中真正来源于细胞的信号占比,是评估背景 RNA 和空液滴影响程度的重要指标;较高的数值通常表示大多数 Reads 来自真实细胞,而数值偏低则提示存在较多背景 RNA、空液滴或细胞破裂释放的游离 RNA。
Mean Reads per Cell (平均每个细胞的 Reads 数,RNA)
- 定义: 在被识别为有效细胞的 Barcode 中,平均每个细胞分配到的测序 Reads 数量。
- 计算方法: 将所有来源于有效细胞 Barcode 的 Reads 数量相加,并除以有效细胞的总数。
- 解读: 该指标反映测序深度在单细胞层面的分配情况,数值越高通常表示单个细胞获得的测序信息越充分,有助于提高基因表达定量的稳定性,但需结合测序饱和度和文库复杂度综合判断。
Median Genes per Cell (中位基因数,RNA)
- 定义: 有效细胞中,每个细胞被检测到的基因数量的中位数。
- 计算方法: 统计所有有效细胞中每个细胞具有至少一个 UMI 计数的基因数,并对该分布取中位数。
- 解读: 该指标反映 RNA 文库的检测灵敏度和复杂度,数值越高通常表示文库质量较好或测序深度较高,从而能够检测到更多低表达基因。
Median UMI Counts per Cell (每个细胞的 UMI 数中位数,RNA)
- 定义: 在被识别为有效细胞的 Barcode 中,每个细胞检测到的唯一 UMI 数量的中位数。
- 计算方法: 对所有有效细胞分别统计其唯一 UMI 数量(每个 UMI 仅计数一次),并对该分布取中位数。
- 解读: 该指标反映单细胞层面转录分子捕获数量的典型水平,数值越高通常表示单个细胞中可用于定量的转录分子信息越丰富,但需结合测序深度和文库复杂度综合评估。
Total Genes Detected (检测到的总基因数,RNA)
- 定义: 在所有被识别为有效细胞的 Barcode 中,检测到的不同基因的总数量。
- 计算方法: 在所有有效细胞中,对经 UMI 去重后的基因进行汇总统计,得到被检测到的不同基因数量。
- 解读: 指标反映转录组文库在整体层面对基因表达的覆盖广度,数值越高通常表示在当前数据中能够检测到的基因种类越丰富,但需结合细胞数量、测序深度及样本类型综合解读。
Sequencing Depth and Saturation Analysis(测序深度与饱和度分析,RNA)
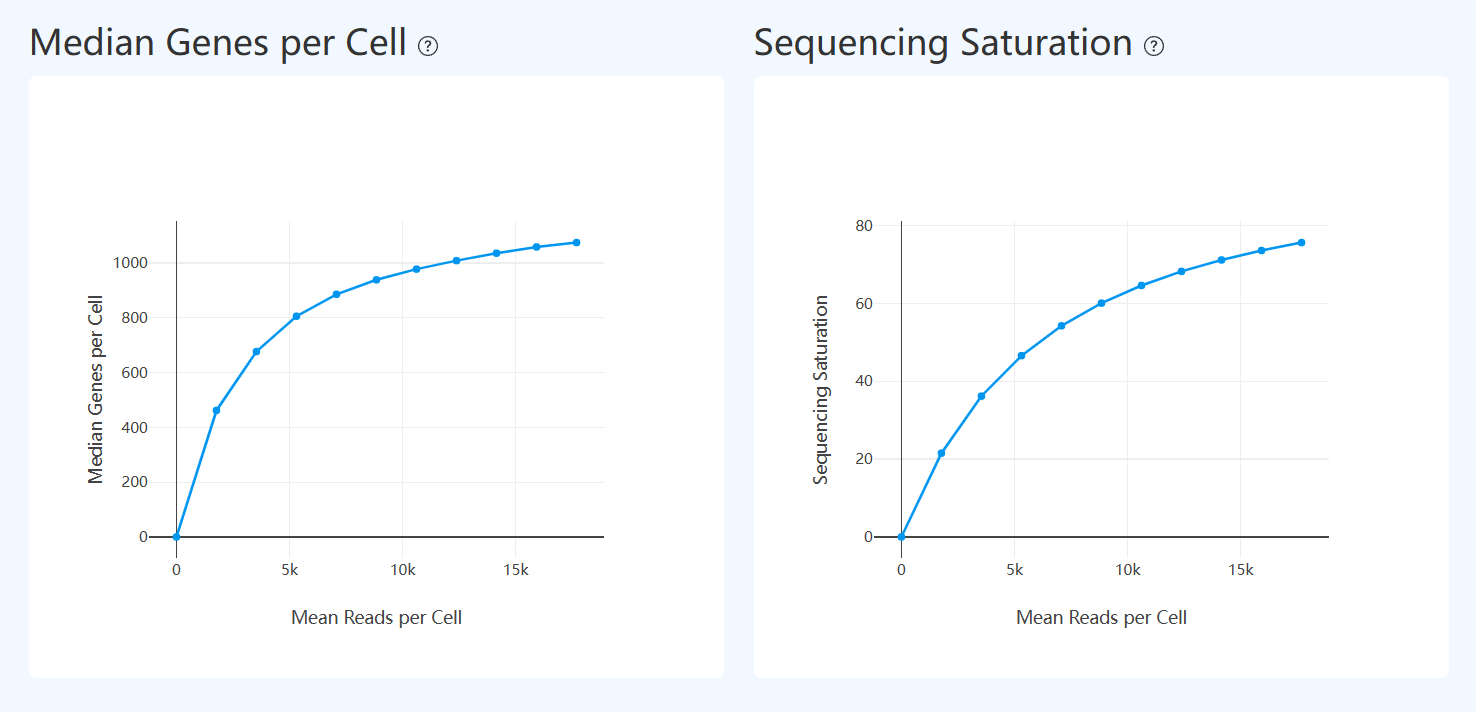
Median Genes per Cell (测序深度与基因检测能力,RNA)
- 定义: 基于对 RNA 测序数据进行下采样处理,展示在不同 Mean Reads per Cell 条件下,单细胞 Median Genes per Cell 的变化关系。横轴表示每个细胞的平均测序 reads 数,纵轴表示对应条件下检测到的基因数中位数。
- 解读: 该图用于评估测序深度对单细胞转录组基因检测能力的影响及其饱和趋势。一般而言,随着每个细胞分配的测序 reads 增加,能够检测到的基因数随之上升;当曲线逐渐趋于平缓时,说明新增测序 reads 对基因检测数量的增益开始减弱。该趋势可用于判断当前测序深度是否已接近文库复杂度上限,并为是否需要进一步加深测序提供参考。
Sequencing Saturation(测序深度与重复率关系,RNA)
- 定义: 基于对 RNA 测序数据进行下采样处理,展示在不同测序深度条件下,Sequencing Saturation 随测序数据投入变化的趋势。横轴表示下采样后对应的表达信息量水平(以单细胞层面的基因检测情况为参照),纵轴表示测序饱和度,用于反映重复测序分子的比例。
- 解读: 该图用于评估测序深度与文库复杂度之间的关系。随着测序深度增加,测序饱和度通常逐步升高,表示新增 reads 中重复分子的比例增加;当曲线趋于平缓时,说明进一步增加测序深度对获取新的转录分子信息的边际收益有限。该趋势可与基因数或 UMI 数的下采样曲线结合解读,用于综合判断当前测序深度是否已接近文库复杂度上限。
Methylation Metrics (甲基化指标)
本部分展示单细胞甲基化文库在测序质量、转化效率、比对情况及甲基化水平等方面的核心质控指标,用于评估甲基化数据的可靠性、有效性及其下游分析可行性。
Sequencing (测序质量,MET)
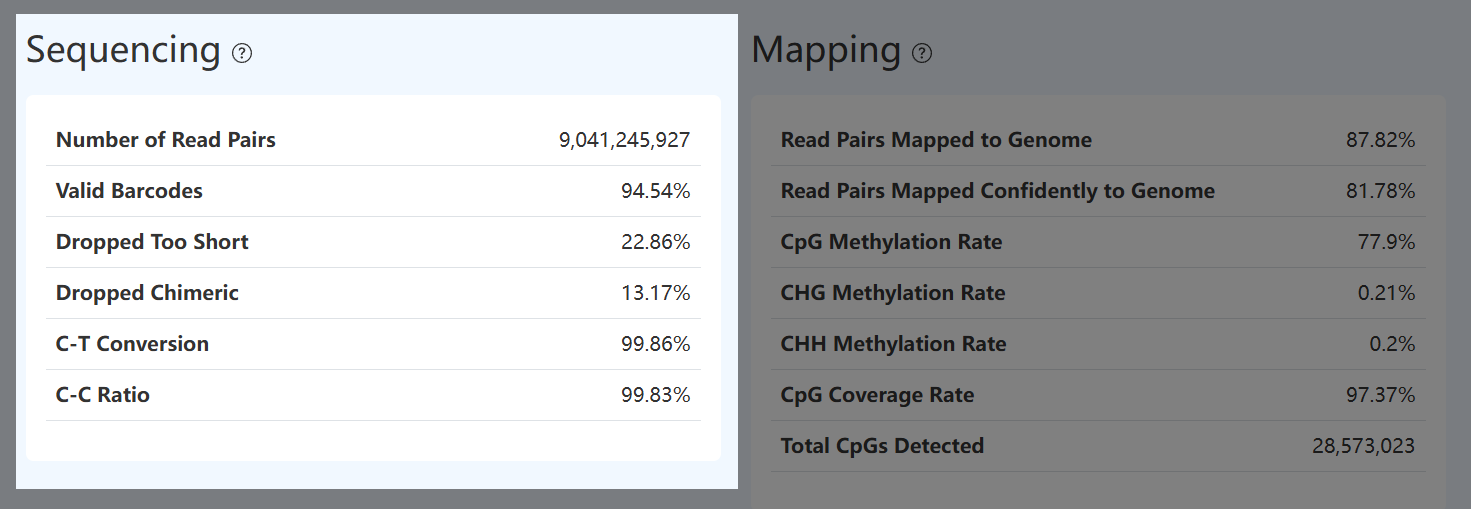
Number of Read Pairs (读取对数量,MET)
- 定义: fastp 质控后保留下来的甲基化测序 reads pairs 的总数量。
- 计算方法: 对甲基化测序原始数据进行 fastp 质控后,统计成功保留并形成配对的 reads pairs 数量。
- 解读: 该指标反映甲基化测序的总体数据产出规模,其数值需结合有效 Barcode 比例、基因组比对率及细胞内 Reads 占比综合评估,判断测序数据是否有效转化为可用于下游分析的信息。
Valid Barcode (有效 Barcode 比例,MET)
- 定义: 在 fastp 质控后的甲基化测序 Reads 中,Barcode 序列能够匹配至细胞白名单的 reads pairs 所占比例。
- 计算方法: 统计 Barcode 序列在纠错前直接匹配或在允许碱基纠错后匹配至白名单的 reads pairs 数量,并除以 Number of Read Pairs。
- 解读: 该指标反映甲基化文库中细胞 Barcode 的保留和识别情况;当数值偏低时,除建库过程中酶切或连接效率不足导致 Barcode 片段受损或丢失外,还可通过检查 fastp 结果或 Reads 起始碱基组成中是否存在异常偏高的 C 碱基比例,辅助判断文库是否存在结构异常或转化过程相关问题。
Dropped Too Short (过短序列过滤比例,MET)
- 定义: 在去除接头(adapter)后,由于 R1(20 bp)或 R2(60 bp)长度不足而被丢弃的 reads pairs 所占比例。
- 计算方法: 统计 adapter 去除后因任一端序列长度低于最小阈值而被过滤的 reads pairs 数量,并除以 Number of Read Pairs。
- 解读: 该指标反映甲基化文库中插入片段长度及结构完整性情况;比例偏高通常提示插入片段过短、接头残留较多或建库过程中发生片段降解,从而导致有效 reads pairs 无法保留。
Dropped Chimeric (嵌合体过滤比例,MET)
- 定义: 在满足结构要求(包含有效 Barcode,且 R1 ≥ 20 bp、R2 ≥ 60 bp)的 reads pairs 中,因 CH 上下文甲基化位点数量异常而被判定为嵌合体或转化假象并被过滤掉的 reads pairs 所占比例。
- 计算方法: 在通过 Barcode 校验及最小长度过滤的 reads pairs 中,依据 CH 位点甲基化模式及预设过滤规则,统计因 CH-context 甲基化异常而被移除的 reads pairs 数量,并除以满足上述结构要求的 reads pairs 总数。
- 解读: 该指标反映甲基化数据中因实验或扩增过程引入的嵌合体及异常转化 reads 的比例;数值偏高通常提示文库中存在较多非真实甲基化信号,可能与 PCR 嵌合体、转化异常或文库结构异常有关。
C-T Conversion (C–T 转化率,MET)
- 定义: 甲基化处理过程中,未甲基化胞嘧啶(C)被成功转化为胸腺嘧啶(T)的效率。
- 计算方法: 在结构完整、可用于甲基化判定的 reads pairs 中(即同时正确识别 7F、17L 和 ME 结构序列的 reads pairs),基于 Forward 链统计 17L/ME 上已知为未甲基化的 C 位点 中被转化为 T 的比例,作为 C–T 转化率。
- 解读: 该指标是评估甲基化文库质量的核心指标之一,转化率偏低会导致未甲基化的 C 被误判为甲基化,从而引入假阳性结果,通常需要结合 CHG/CHH 甲基化水平一并评估。
C-C Ratio (C检测准确率,MET)
- 定义: 甲基化转化过程中,已甲基化的胞嘧啶(C)被正确保持为 C 的比例,用于评估转化过程的特异性。
- 计算方法: 在结构完整、可用于甲基化判定的 reads pairs 中,基于 Reverse 链 统计 17L/ME 上已知为甲基化的 C 位点 中,未被错误转化为 T、仍保持为 C 的比例,作为 C–C 保持率。
- 解读: 该指标是评估甲基化处理特异性的核心质量指标;数值偏低通常提示转化过程过度,已甲基化的 C 被错误转化,从而导致甲基化信号丢失并引入假阴性结果,需与 C–T 转化率联合解读。
Mapping (比对情况,MET)
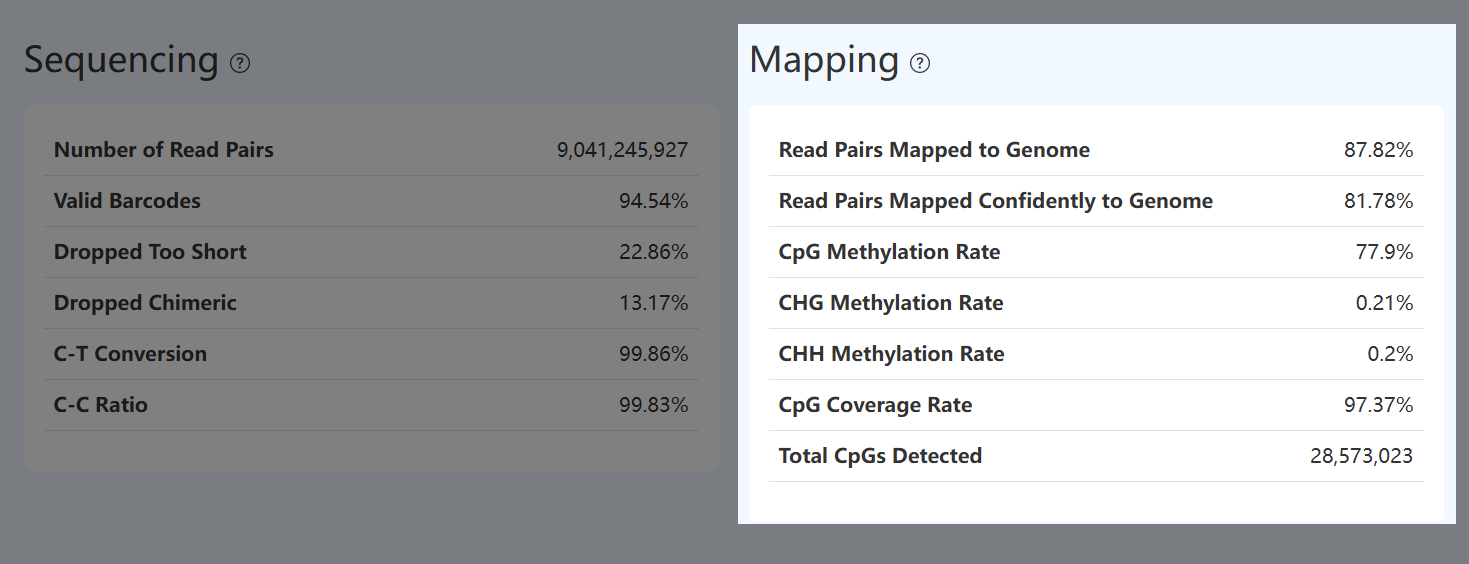
Read Pairs Mapped to Genome (基因组比对率,MET)
- 定义: 在甲基化测序数据中,参与基因组比对的 reads pairs 中,成功比对到参考基因组的 reads pais 所占比例。
- 计算方法: 在完成 Barcode 校验、过短 reads 过滤及 CH 异常 reads pairs 过滤后,统计实际参与 Bismark 比对的 reads pairs 数量;其中成功比对到参考基因组的 reads pairs 数量除以参与比对的 reads pairs 总数,得到基因组比对率。该指标基于 Bismark 比对报告计算得到。
- 解读: 该指标反映甲基化测序数据在有效 reads 层面的的基因组映射效率,当比对率偏低时,可能与样本或文库中存在污染、接头去除不充分,或插入片段长度过大,从而影响比对效率有关,应结合唯一比对率及有效 Barcode 比例综合判断。
Read Pairs Mapped Confidently to Genome (唯一比对率,MET)
- 定义: 在参与基因组比对的甲基化 reads pairs 中,能够以高置信度、唯一方式比对到参考基因组的 reads pairs 所占比例。
- 计算方法: 基于完成预处理并实际参与 Bismark 比对的 reads pairs,统计其中仅存在一个最佳比对位置的 reads pairs 数量,并除以参与比对的 reads pairs 总数;该指标根据 Bismark 比对报告计算得到。
- 解读: 该指标反映甲基化测序数据的比对确定性和定位可靠性。唯一比对率偏低时,除可能存在污染或文库结构异常外,也可能与插入片段过长或重复序列比例较高有关,需结合总体比对率和后续甲基化覆盖指标综合评估。
CpG Methylation Rate (CpG 甲基化率,MET)
- 定义: 在全基因组范围内,CpG 位点的平均甲基化水平。
- 计算方法: 基于高置信度唯一比对到参考基因组的 reads pairs,在 CpG 上下文(CpG context) 中统计被判定为甲基化的 C 的总数,并除以该上下文中检测到的总信号数 (C + T),该指标根据 Bismark 比对与甲基化统计报告计算得到。
- 解读: 该指标反映样本的全局 CpG 甲基化状态,显著偏离该范围可能提示样本类型异常、转化效率问题或文库质量异常,需结合 C–T 转化率与 C–C 保持率综合判断。
CHG Methylation Rate (非 CpG 甲基化率 – CHG,MET)
- 定义: 在非 CpG 上下文(CHG)中,胞嘧啶(C)的平均甲基化水平。
- 计算方法: 基于高置信度唯一比对到参考基因组的 reads pairs,在 CHG 上下文(CHG context) 中统计被判定为甲基化的 C 的总数,并除以该上下文中检测到的总信号数 (C + T);该指标根据 Bismark 比对与甲基化统计报告计算得到。
- 解读: 该指标反映样本中非 CpG 位点的甲基化背景水平,在人和小鼠的大多数体细胞组织中通常极低(一般 < 1%);若该值升高,可能提示甲基化转化不完全,或样本中包含具有非 CpG 甲基化特征的特殊细胞类型(如神经元),需结合 C–T 转化率及 CpG 甲基化率综合判断。
CHH Methylation Rate (非 CpG 甲基化率 – CHH,MET)
- 定义: 在非 CpG 上下文(CHH)中,胞嘧啶(C)的平均甲基化水平。
- 计算方法: 基于高置信度唯一比对到参考基因组的 reads pairs,在 CHH 上下文(CHH context) 中统计被判定为甲基化的 C 的总数,并除以该上下文中检测到的总信号数 (C + T);该指标根据 Bismark 比对与甲基化统计报告计算得到。
- 解读: 与 CHG 类似,该指标反映样本中非 CpG 位点的甲基化背景水平,在人和小鼠的大多数体细胞组织中通常极低(一般 < 1%);若该值升高,可能提示甲基化转化不完全,或样本中包含具有非 CpG 甲基化特征的特殊细胞类型(如神经元),需结合 C–T 转化率及 CpG 甲基化率综合判断。
CpG Coverage Rate (CpG 覆盖率,MET)
- 定义: 在参考基因组中,理论 CpG 位点被实际检测到的比例。
- 计算方法: 将所有细胞的甲基化数据进行汇总(视为 bulk),统计被至少一次检测到的 CpG 位点数量,并除以参考基因组中理论 CpG 位点的总数(计算时过滤线粒体染色体及未定位的 contig),得到 CpG 覆盖率。
- 解读: 该指标反映甲基化测序在全基因组层面的覆盖广度,数值越高表示检测到的 CpG 位点范围越广,有助于支持更全面和稳定的甲基化模式分析。
Total CpGs Detected (检测到的总 CpG 位点数,MET)
- 定义: 在合并所有细胞的甲基化数据后,至少被一次有效检测到的不同 CpG 位点总数。
- 计算方法: 将所有细胞的甲基化数据进行汇总(视为 bulk),统计被至少 1 条有效比对 reads 覆盖并通过甲基化判定的唯一 CpG 位点数量。
- 解读: 该指标反映甲基化测序实际捕获到的 CpG 位点空间规模,其数值受测序深度、细胞数量及参考基因组中 CpG 位点分布影响,不同物种之间的理论 CpG 总数差异较大,需结合 CpG 覆盖率综合解读。
Cells (甲基化细胞统计,MET)
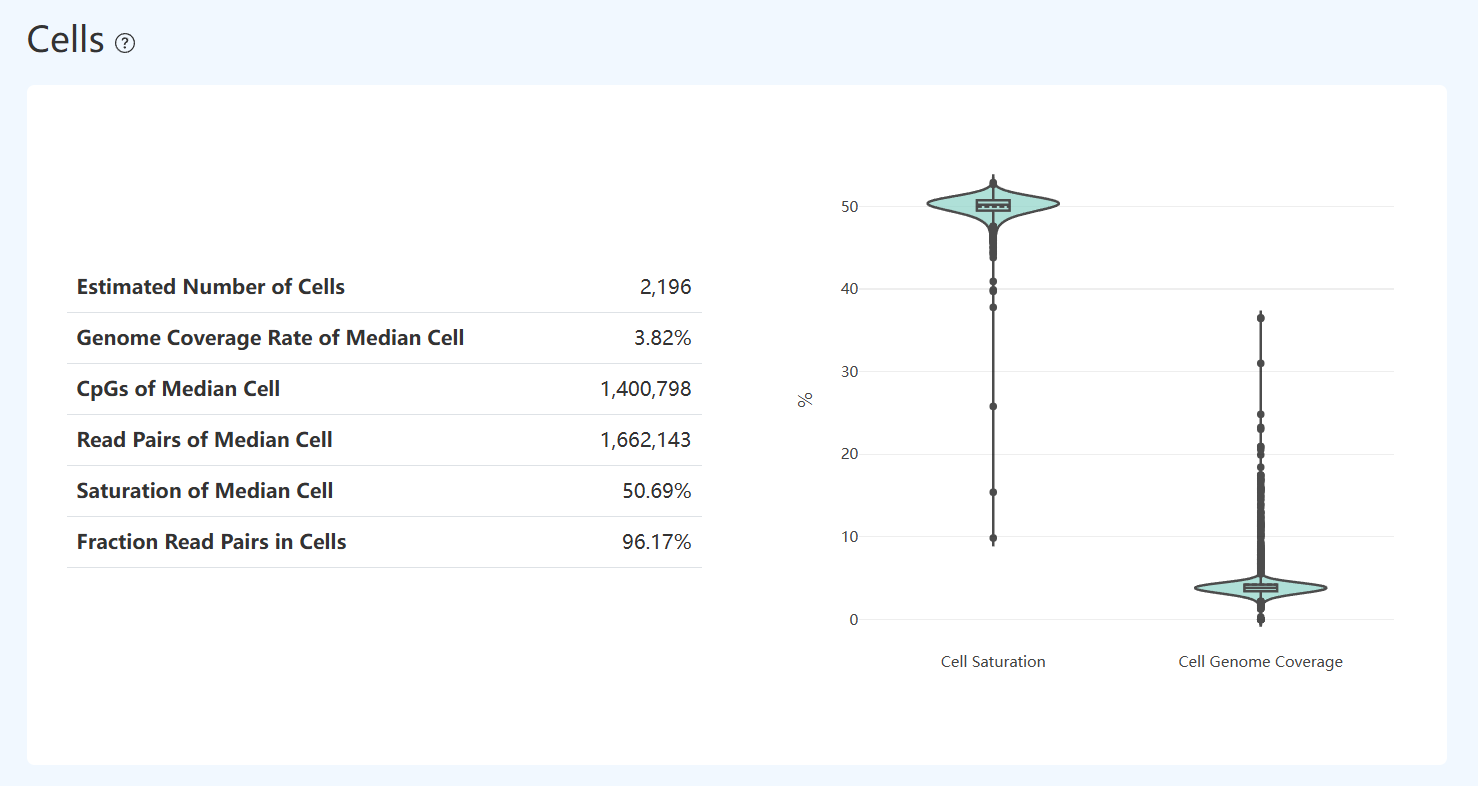
Genome Coverage Rate of Median Cell (中位细胞基因组覆盖度,MET)
- 定义: 在所有有效细胞中,基因组覆盖度处于中位水平的细胞所对应的基因组覆盖比例。
- 计算方法: 分别计算每个细胞被检测到的基因组碱基数占参考基因组总大小的比例,按覆盖度对细胞排序后,选取位于中位的细胞,其覆盖比例即为该指标。
- 解读: 该指标反映单个代表性细胞在基因组层面的信息覆盖程度;由于单细胞甲基化数据具有高度稀疏性,该指标可直观展示“典型”单个细胞可获得的基因组信息范围。
CpGs of Median Cell (中位细胞 CpG 数,MET)
- 定义: 基因组覆盖度处于中位水平的细胞中,被检测到的 CpG 位点数量。
- 计算方法: 根据每个细胞的基因组覆盖度对有效细胞排序,选取覆盖度位于中位的细胞,并统计该细胞中至少被 1 条通过过滤并成功比对的 reads 覆盖、且产生有效甲基化判定的 CpG 位点总数(正负链分别计数)。
- 解读: 该指标反映单个代表性细胞在甲基化数据中的信息量水平,数值越高表示该细胞在其覆盖范围内检测到的 CpG 位点越多,单细胞层面的甲基化信息相对更丰富。
Read Pairs of Median Cell (中位细胞唯一比对的读取对数量,MET)
- 定义: 在甲基化文库中,基因组覆盖度处于中位水平的有效细胞内,能够唯一且高置信度比对到参考基因组的 reads pairs 数量。
- 计算方法: 先根据每个有效细胞的基因组覆盖度对细胞进行排序,选取覆盖度位于中位的细胞;在该细胞对应的 reads pairs 中,统计具有 unique best alignment、且成功比对到参考基因组的 reads pairs 数量;该统计基于 Bismark 比对报告获得。
- 解读: 该指标反映单个代表性细胞中,可用于甲基化分析的高质量测序数据规模;数值越高,通常表示该细胞获得的有效甲基化测序信息越多,但需结合该细胞的基因组覆盖度和 CpG 数量综合解读。
Saturation of Median Cell (中位细胞饱和度,MET)
- 定义: 针对基因组覆盖度处于中位水平的细胞,评估其甲基化测序数据在 DNA 分子层面是否已接近饱和。
- 计算方法: 选取基因组覆盖度位于中位的细胞,在其覆盖的 CpG 位点上,统计去重后的唯一 DNA 分子数,并除以用于检测这些位点的总 reads 数,再以 Saturation = 1 −(唯一 DNA 分子数 / 总 reads 数)计算得到该细胞的测序饱和度。
- 解读: 该指标用于反映该代表性细胞中测序深度与文库复杂度的关系。一般而言,饱和度较高表示 reads 的重复比例更高,继续加深测序对新增 DNA 分子信息的提升相对有限;饱和度较低则说明仍有较多未被充分采样的分子,增加测序深度仍可能带来更多可用于甲基化判定的信息。
Fraction Read Pairs in Cells (细胞内 Read Pairs 占比,MET)
- 定义: 在甲基化测序数据中,成功唯一比对到参考基因组的 reads pairs 中,来源于被识别为有效细胞 Barcode 的 reads pairs 所占比例。
- 计算方法: 在所有 唯一且高置信度比对到参考基因组的 reads pairs 中,统计属于有效细胞 Barcode 的 reads pairs 数量,并除以唯一比对 reads pairs 的总数;该指标基于比对与细胞识别结果计算得到。
- 解读: 该指标反映甲基化测序数据中真正来源于有效细胞的信号占比,是评估背景噪音和无效 Barcode 影响程度的重要指标;在采用 DD-MET3 双标签体系的实验中,由于 Barcode 匹配与识别过程本身存在一定损耗,该指标通常相对较低,需结合有效 Barcode 比例和比对率综合判断。
Cell Saturation & Genome Coverage Distribution(细胞饱和度与基因组覆盖度分布,MET)
- 定义: 右侧小提琴图分别展示在所有有效细胞中,按细胞逐一计算得到的 Cell Saturation 与 Cell Genome Coverage 的分布情况;箱线表示中位数及四分位范围,散点代表单个细胞。
- 解读: 该图用于快速查看细胞间在测序重复程度(Cell Saturation)与基因组覆盖广度(Cell Genome Coverage)上的一致性与差异程度。
- 分布更集中: 通常表示细胞间差异相对较小,整体一致性较好;
- 分布较发散或离群点较多: 表示存在差异较大的细胞,可结合该细胞的 reads pairs、CpG 数及过滤相关指标进一步评估。
Single-cell Methylation Level and Information Content(单细胞甲基化水平与信息量,MET)
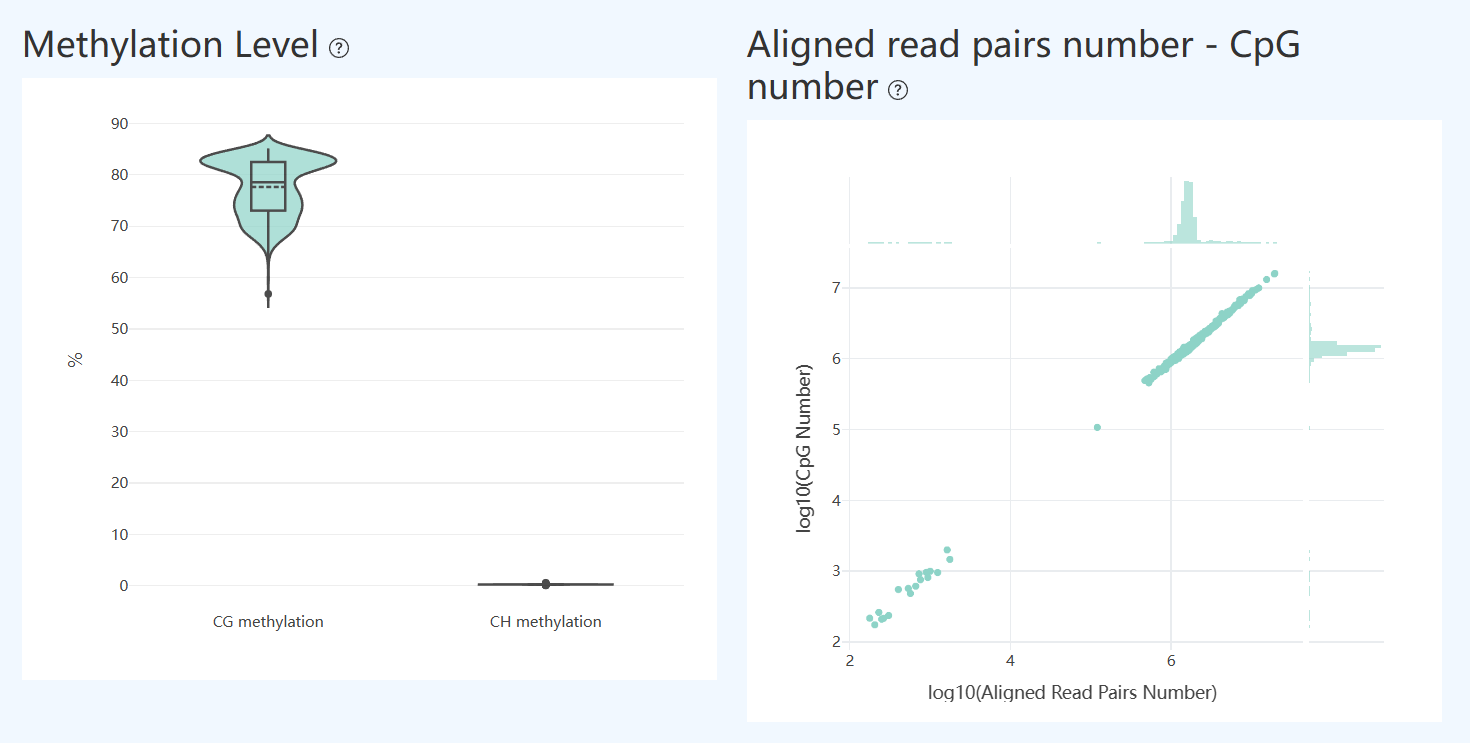
Methylation Level(全局甲基化水平分布,MET)
- 定义: 小提琴图用于展示单细胞层面 CG(CpG) 与 CH(非 CpG,包含 CHG/CHH) 的甲基化率的分布情况;箱线表示中位数及四分位范围,散点代表单个细胞。
- 解读: 该图用于评估样本的整体甲基化状态及细胞间一致性。通常情况下,CG 甲基化在主要细胞群体中应呈相对集中的分布,反映全局甲基化水平较为稳定;CH 甲基化一般处于较低背景水平。
- 异常情形 1 (CH 升高): 若 CH 分布整体抬高或出现明显长尾,可能提示转化效率不足或背景噪音升高(亦可能与特定细胞类型的生物学特征相关);
- 异常情形 2 (CG 发散): 若 CG 分布明显发散或存在较多离群细胞,常见于细胞质量不均或覆盖不足,建议结合单细胞覆盖度及有效 CpG 位点数进一步核查。
Aligned read pairs number – CpG number(唯一比对 reads 对数与 CpG 检测数关系,MET)
- 定义: 散点图展示单细胞层面唯一且高置信度比对到参考基因组的 reads pairs 数量(X 轴)与该细胞检测到的 CpG 位点数(Y 轴)之间的关系;图中采用对数坐标,并附带边缘分布以展示两项指标的整体分布特征。
- 解读: 该图用于评估测序数据在单细胞层面向甲基化信息量的转化效率。
- 正常趋势: 一般而言,唯一比对 reads pairs 数量与 CpG 检测数应呈现稳定的正相关关系,表明测序数据能够有效支持 CpG 位点的检测。
- 异常偏离: 若存在明显偏离主趋势的细胞,通常提示细胞间质量差异、重复或低复杂度 reads 占比较高,或比对及甲基化判定有效性不足。此类细胞可结合覆盖度、饱和度及过滤比例等指标进一步评估。