单细胞空间转录组质控报告说明文档
本文档详细说明了 SeekSpace Tools 软件输出的空间转录组质控报告中各项指标与图表的定义、计算方法与解读指南,旨在帮助用户理解测序数据与文库质量状况,并为后续数据分析提供参考依据。
报告主要分为两个部分:
- Spatial (测序与空间定位相关):包含 Summary、RNA Library、RNA Mapping、Spatial Library、深度/饱和度曲线、Barcode rank 等质控汇总与诊断图。
- Analysis (分析结果相关):包含降维聚类(t-SNE)、组织空间分布、marker 基因展示、按簇的差异基因等。
Joint Metrics (联合分析指标)
此部分展示空间转录组与单细胞特征相结合后的核心质控指标,用于整体评估切片上的有效细胞与基因捕获情况。
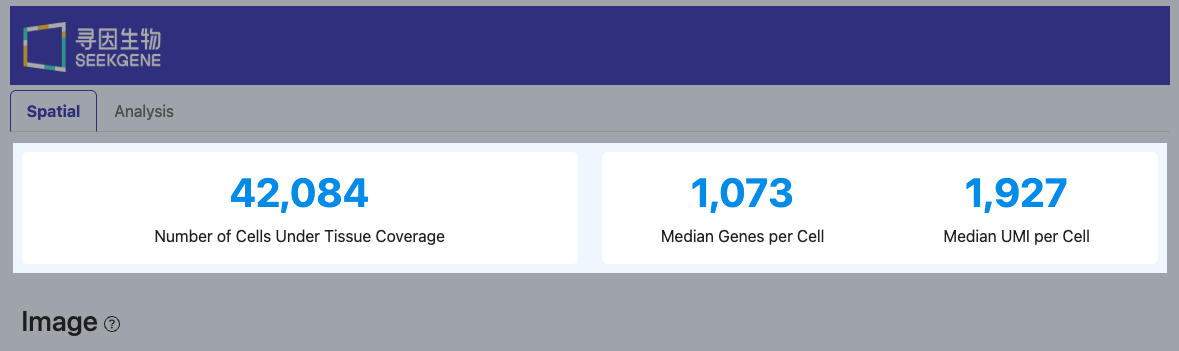
Number of Cells Under Tissue Coverage(组织覆盖内有效细胞数)
(该指标与 Summary 模块中的 “Number of Cells Under Tissue Coverage” 定义一致)
- 定义: 最终落在组织覆盖区域内的有效细胞 barcode 数量。
- 计算方法: 在完成组织区域判定后,仅保留具有唯一中心点且位于组织覆盖区域内的细胞 barcode。
- 解读: 该指标直接对应“真正用于计数与下游分析”的细胞规模。
- 若该值远小于 Number of Cells with Unique Center:重点排查组织区域识别、图像/点位错位(Adjustment)、以及切片组织是否完整覆盖捕获区域。
Median Genes per Cell(每个细胞中位基因数)
(该指标与 Summary 模块中的 “Median Genes per Cell” 定义一致)
- 定义: 组织覆盖内的有效细胞中,每个细胞被检测到的不同基因数量的中位数。
- 计算方法: 汇总组织覆盖内所有细胞的基因表达矩阵,统计每个细胞中具有至少 1 个 UMI 计数的基因数量,并对全体细胞的该数值分布取中位数。
- 解读: 该指标是评估 RNA 文库检测灵敏度与单细胞信息复杂度的核心标准。数值越高,通常表示文库质量越好、测序深度越充足,能捕获到更多中低丰度的转录本。
- 若细胞数充足但该指标偏低:建议按以下顺序排查:(1) 测序深度是否不足(查看 Mean Reads per Cell,或下采样曲线是否未达平台期);(2) RNA 是否存在严重降解(结合 Mapping 模块的比对率、Too Short 比例及外显子/内含子分布异常进行判断);(3) 细胞本身质量较差或环境 RNA 污染严重(查看 Fraction Reads in Cells 是否降低)。
Median UMI per Cell(每个细胞中位 UMI)
(该指标与 Summary 模块中的 “Median UMI per Cell” 定义一致)
- 定义: 组织覆盖内的有效细胞中,每个细胞所包含的 UMI 总数的中位数。
- 计算方法: 汇总组织覆盖内所有细胞的表达矩阵,统计每个细胞分配到的总 UMI 数量,并对全体细胞的该数值分布取中位数。
- 解读: 该指标反映单个有效细胞捕获到的整体转录本分子丰度,是衡量文库转化效率和整体表达水平的直观反映。通常与 Median Genes per Cell 呈正相关趋势。
Interactive Spatial Visualization(交互式空间可视化)
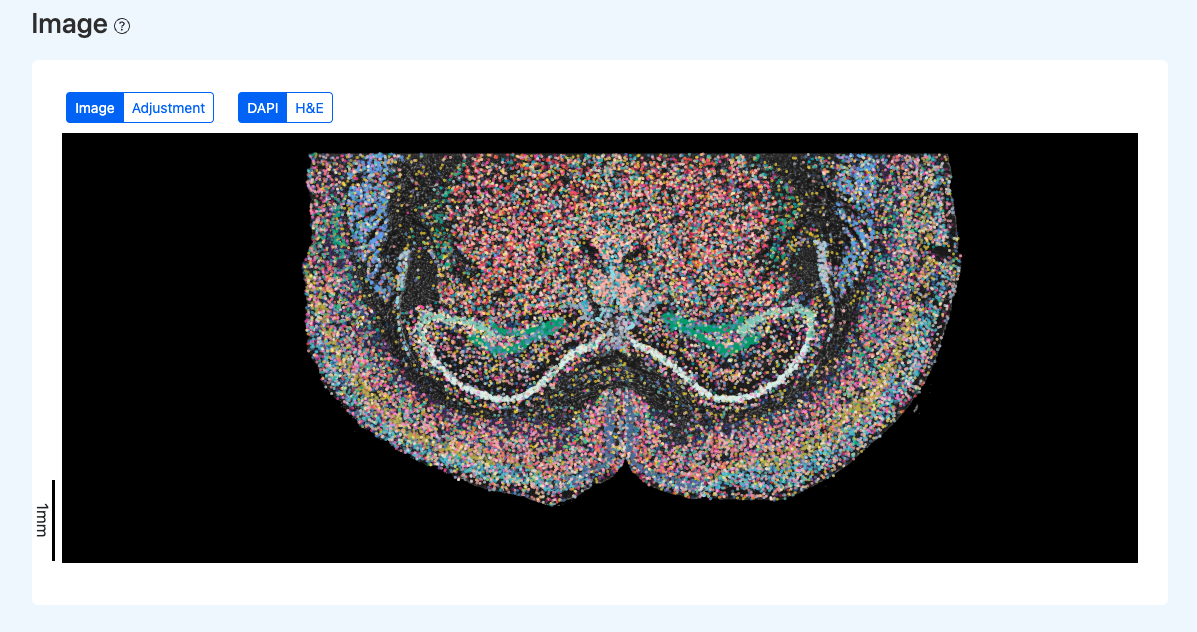
图示说明:
- 点击 H&E 可切换底片为 HE 图像(若报告包含 HE)。
- 点击 Adjustment 可进入图像与细胞点位的手动校准界面,详见“手动校准(Adjustment)”章节。
Summary(总体概览)
本部分汇总了空间转录组测序数据的核心质控指标,旨在帮助用户宏观评估实验整体质量、细胞产出规模及数据信息丰富度。建议优先通过此模块完成对数据可用性的综合判定,随后结合 RNA 与 Spatial 模块进行更深度的指标诊断与异常溯源。
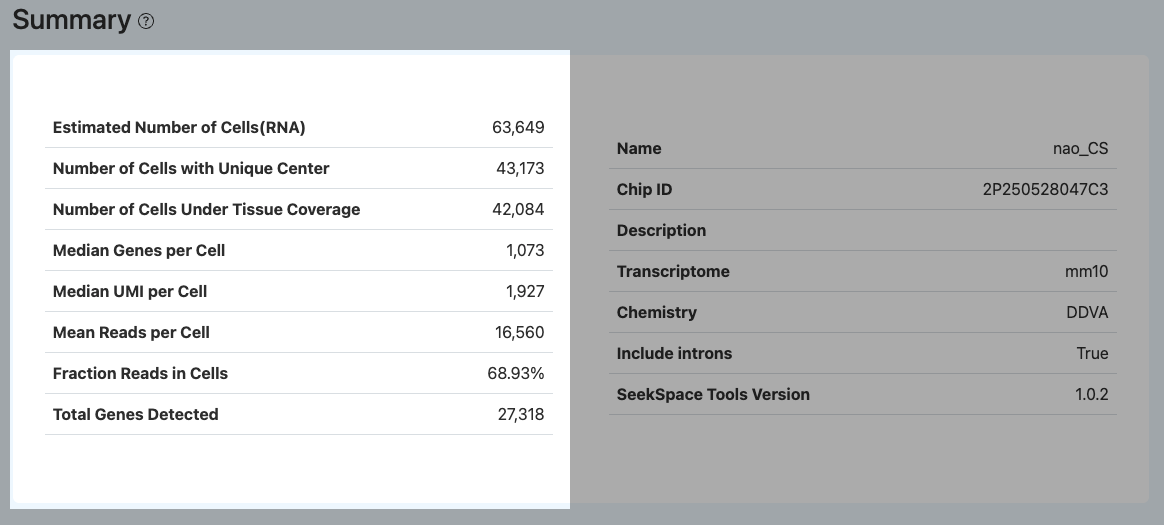
Estimated Number of Cells (RNA)(RNA 文库细胞数)
- 定义: RNA 文库中被判定为有效细胞的 Barcode 数量。
- 计算方法: 基于每个 Barcode 的 RNA UMI 计数分布,默认通过
forceCell方法筛选出有效细胞。首先选取 UMI 总数排名前 80k 的 Barcode 作为候选群体,随后过滤掉 UMI < 200 的低质量 Barcode,最终保留的即为 RNA 文库中的有效细胞数。 - 解读: 该指标反映 RNA 文库可用于后续分析的细胞规模。其数值受样本起始量、细胞活性、裂解效率、测序深度以及细胞判定阈值等多种因素影响。
- 若该数值明显偏低:建议优先排查组织切片质量、测序数据量(结合 Number of Reads 与 Mean Reads per Cell),以及 Barcode 测序质量(Valid Barcode、Q30)。
Number of Cells with Unique Center(具唯一中心的细胞数)
- 定义: 在空间定位流程中,能够成功计算并分配到唯一空间中心坐标(Cell Center)的有效细胞 Barcode 数量。
- 计算方法: 针对被判定为有效细胞的 Barcode,系统会结合其对应的空间 Barcode(Spatial Barcode)在芯片上的物理分布与聚集特征来估算细胞中心。若该分布能拟合出唯一明确的中心点坐标,则该细胞被计入此项。
- 解读: 该指标反映细胞在空间芯片上定位的准确性。理想情况下,该数值应达到 Estimated Number of Cells (RNA) 的 50% 以上。
- 若该数值比例偏低:通常提示空间 Barcode 定位存在模糊或散乱。建议核查 Spatial 模块中的 Accurate Spatial UMIs 和 Mean Spatial UMIs per Cell 指标,排查是否存在空间信号扩散等异常情况。
Number of Cells Under Tissue Coverage(组织覆盖内有效细胞数)
- 定义: 最终落入被识别为组织覆盖区域(Tissue Covered Area)内部的、且具备唯一中心点的有效细胞 Barcode 数量。
- 计算方法: 在完成图像配准与组织区域判定后,系统从“具唯一中心的细胞”集合中,进一步筛选出其中心坐标位于组织多边形轮廓内的细胞。
- 解读: 该指标直接代表了“真正能够用于空间表达定量与下游空间特征分析”的细胞真实规模。
- 若该数值远低于 Number of Cells with Unique Center:说明大量判定出的细胞散落在了组织区域之外。需重点排查组织轮廓识别是否准确、图像与空间点阵是否存在严重错位(可进入 Adjustment 核查),以及切片组织是否未能完整覆盖芯片的有效捕获区。
Median Genes per Cell(每个细胞中位基因数)
- 定义: 组织覆盖内的有效细胞中,每个细胞被检测到的不同基因数量的中位数。
- 计算方法: 汇总组织覆盖内所有细胞的基因表达矩阵,统计每个细胞中具有至少 1 个 UMI 计数的基因数量,并对全体细胞的该数值分布取中位数。
- 解读: 该指标是评估 RNA 文库检测灵敏度与单细胞信息复杂度的核心标准。数值越高,通常表示文库质量越好、测序深度越充足,能捕获到更多中低丰度的转录本。
- 若细胞数充足但该指标偏低:建议按以下顺序排查:(1) 测序深度是否不足(查看 Mean Reads per Cell,或下采样曲线是否未达平台期);(2) RNA 是否存在严重降解(结合 Mapping 模块的比对率、Too Short 比例及外显子/内含子分布异常进行判断);(3) 细胞本身质量较差或环境 RNA 污染严重(查看 Fraction Reads in Cells 是否降低)。
Median UMI per Cell(每个细胞中位 UMI)
- 定义: 组织覆盖内的有效细胞中,每个细胞所包含的 UMI 总数的中位数。
- 计算方法: 汇总组织覆盖内所有细胞的表达矩阵,统计每个细胞分配到的总 UMI 数量,并对全体细胞的该数值分布取中位数。
- 解读: 该指标反映单个有效细胞捕获到的整体转录本分子丰度,是衡量文库转化效率和整体表达水平的直观反映。通常与 Median Genes per Cell 呈正相关趋势。
Mean Reads per Cell(平均每个细胞的 Reads 数)
- 定义: 反映平均分配给每个组织覆盖内有效细胞的测序数据量规模。
- 计算方法: RNA 文库总有效 Reads 数(Number of Reads)除以组织覆盖内有效细胞数(Number of Cells Under Tissue Coverage)。
- 解读: 该指标并非指细胞内实际真实捕获的 Reads 绝对数量,而是用于宏观评估当前整体测序深度是否充裕。增加该数值通常有助于提升 Median Genes/UMI。
- 需注意:该指标并非越高越好。若 Mean Reads per Cell 已经很高,但 Median Genes/UMI 的提升趋于停滞,且 Sequencing Saturation(测序饱和度)接近饱和,则提示文库复杂度已达极限或重复序列测序比例过高,继续增加测序量带来的信息增益有限。
Fraction Reads in Cells(细胞内 reads 占比)
- 定义: 在成功比对到参考基因组且分配到已知基因注释区域的 Reads 中,最终归属于组织覆盖内有效细胞的 Reads 所占的比例。
- 计算方法: 将分配给组织覆盖内细胞的基因计数 Reads 总和,除以所有成功映射到基因组及转录本注释区域的 Reads 总和。
- 解读: 该指标衡量了测序数据中“有效单细胞信号”的纯度,是评估环境 RNA(Ambient RNA)污染及背景噪声的重要标准。
- 若该比例偏低:通常提示背景游离 RNA 较多。常见原因包括细胞破裂、组织覆盖内的真实有效细胞过少,或者细胞判定阈值(Cell Calling)设置不当(过宽或过严)。
Total Genes Detected(检测到的总基因数)
- 定义: 在所有组织覆盖内的有效细胞群体中,累计检测到的非冗余(不同)基因的总数。
- 计算方法: 合并所有组织覆盖内细胞的表达矩阵,统计在整个群体中至少拥有 1 个 UMI 计数的唯一基因数量。
- 解读: 该指标反映了整个空间样本在当前测序深度下能够覆盖的转录组广度。其数值受物种特性、组织类型复杂度及测序深度影响显著。通常需要结合细胞总数和单细胞水平(per-cell)指标进行综合评估。
- 若有效细胞数充足但总基因数依然偏低:建议重点核查参考基因组/注释文件版本是否匹配、基因组总体比对率是否正常,以及样本本身的 RNA 完整性。
RNA Metrics(转录组指标)
本部分展示单细胞转录组文库的测序质量、比对情况及关键定量指标,用于评估 RNA 数据的整体质量和可用性,并辅助判断其是否满足下游空间分析需求。
RNA Library(测序与 Barcode 质量)

Number of Reads(RNA 文库 Reads 总数)
- 定义: 经过 fastp 质控后保留下来的有效 RNA reads 总数。
- 解读: 该指标反映样本在质控后可用于后续分析的有效测序数据规模,是下游分析的基础输入。通常需结合有效 Barcode 比例、细胞内 Reads 占比及测序饱和度综合判断,避免仅以绝对测序量评估数据有效性。
Valid Barcode(有效 Barcode 比例)
- 定义: RNA 文库的测序 reads 中,Barcode 序列能够准确匹配至已知细胞白名单的 reads 所占比例。
- 计算方法: 统计 Barcode 序列在无需纠错直接匹配,或在允许有限碱基纠错后成功匹配至白名单的 reads 数量,并除以总 Reads 数。
- 解读: 该指标反映 Barcode 合成质量、测序准确性以及白名单匹配情况。一般建议 > 80%(或至少不低于 75%)。当该指标偏低时,建议结合 Barcode Q30 值及细胞内 Reads 占比一并分析,以区分测序质量问题与文库结构或白名单参数不匹配的影响。
Sequencing Saturation(测序饱和度)
- 定义: 用于评估当前测序深度下,文库中转录本分子被重复测序的程度。
- 计算方法: Sequencing Saturation = 1 −(唯一 UMI 数 / 可用于 UMI 计数的 reads 数)。
- 解读: 该指标用于反映测序深度与文库复杂度之间的关系。饱和度升高表示新增测序 reads 中重复分子的比例增加,进一步加深测序对新增分子信息的提升有限;而饱和度较低则说明文库中仍存在较多未被充分采样的分子,增加测序深度仍有助于获得更多新的转录信息。
Too Short(过短 Reads 比例)
- 定义: 在接头(adapter)去除或修剪后,由于序列长度低于最小保留阈值(如 < 60 bp)而被过滤掉的 reads 数量及其占比。
- 解读: 该指标反映 RNA 文库中插入片段长度及结构完整性情况。当该比例偏高时,通常提示接头残留较多、插入片段偏短或文库构建过程中片段发生降解,可能进一步影响后续比对效率和定量分析结果。
Q30 Bases in Barcode / Q30 Bases in UMI(Barcode/UMI 的 Q30 比例)
- 定义: RNA 文库的有效 reads 中,Barcode 和 UMI 序列的测序质量值(Phred score)达到或超过 Q30(即碱基识别错误率 ≤ 0.1%)的碱基所占比例。
- 解读: 该指标反映核心标签序列的测序质量水平。较高的 Q30 比例表示读取准确性较高,有助于提高有效 Barcode 识别率和 UMI 分子去重的准确性;当该指标偏低时,将直接拉低 Valid Barcode 比例,并间接影响细胞识别与转录本定量的可靠性。
RNA Mapping(比对情况)

Reads Mapped to Genome(总体基因组比对率)
- 定义: 在参与基因组比对的 RNA reads 中,成功比对到参考基因组的 reads 所占比例(包含唯一比对与多重比对)。
- 解读: 该指标反映有效 RNA reads 与参考基因组的匹配情况及文库特异性。较高的比对率通常表示样本来源明确且参考基因组选择合理(一般建议 > 80%)。当比对率偏低时,可能与样本污染(如微生物或其他物种)、低复杂度序列比例偏高、Reads 质量差或参考基因组版本不匹配有关。
Reads Mapped Confidently to Genome(唯一比对率)
- 定义: 在参与基因组比对的 RNA reads 中,能够以高置信度唯一比对到参考基因组位置的 reads 所占比例。
- 解读: 该指标反映可用于可靠基因表达定量分析的 reads 比例。唯一比对率偏低时(如低于 50%),可能与重复序列比例较高、测序质量不足或参考基因组不匹配有关,将直接影响下游定量分析的稳定性。
Reads Mapped to Exonic / Intronic / Intergenic Regions(外显子/内含子/基因间区比对率)
- 定义: 在成功比对到参考基因组的 RNA reads 中,比对至已注释外显子、内含子或基因间区域的 reads 分别所占的比例。
- 解读:
- 外显子 (Exonic): 反映成熟 mRNA 信号的富集程度,比例偏高通常表示成熟转录本信号较好,有利于基因表达定量。
- 内含子 (Intronic): 反映未剪接或核内 RNA 信号的占比。比例升高可能与核内 RNA 提取较多、未剪接转录活跃,或分析时开启了
include introns参数有关。 - 基因间区 (Intergenic): 反映 reads 落在已知基因注释区域之外的比例。数值升高需警惕基因组注释不完整、参考版本不匹配、非编码转录本或非特异性比对/污染的影响。
Spatial Metrics(空间文库与定位指标)
此模块主要评估空间测序文库的结构完整性、空间条码(Spatial Barcode)的定位准确性与复杂度,以及空间信号与转录组细胞判定的一致性,是决定空间转录组数据质量及可用性的核心部分。
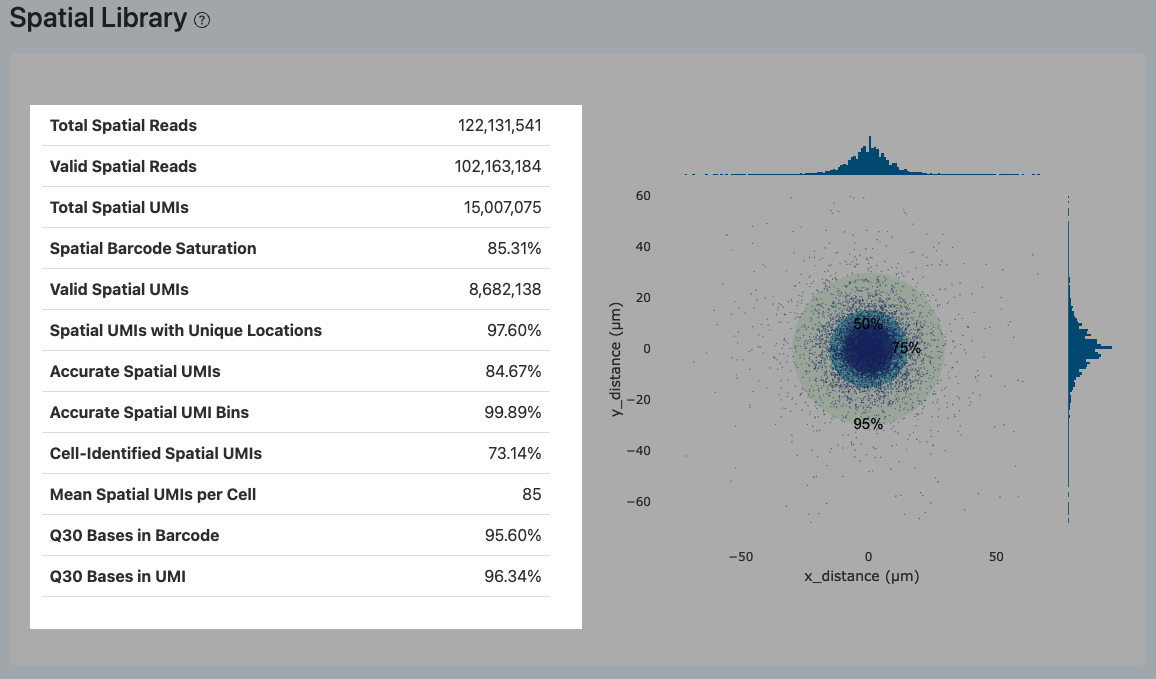
Total Spatial Reads(空间文库总 Reads 数)
- 定义: 空间测序文库中产出的原始 reads 总数。
- 解读: 该指标反映了空间文库的基础测序数据量,是评估测序深度与后续可用数据转化的基数。
Valid Spatial Reads(有效空间 Reads)
- 定义: 满足空间文库规定结构的 reads 数量及其在总 reads 中的占比。
- 计算方法: 统计 Read 1 包含有效细胞 Barcode,且 Read 2 具有至少 32 bp 长度以供成功提取空间 Barcode 的 reads 数量。
- 解读: 该指标直接反映空间文库测序数据的基本结构可用性。
- 若该比例明显偏低:通常提示测序读长设置错误、文库结构不匹配、测序质量较差,或文库构建过程中混入了异常片段。
Total Spatial UMIs(空间 UMI 总数)
- 定义: 从有效空间 reads 中提取,并经过分子标签(UMI)去重后得到的独立空间 UMI 分子总数。
- 计算方法: 对具有相同(Cell Barcode, UMI, Spatial Barcode)组合的 reads 进行去重计数。
- 解读: 反映空间文库的整体捕获信息量。该数值为初步去重后的绝对分子数,但仍需结合后续的定位准确性指标来评估其实际有效的空间信号。
Spatial Barcode Saturation(空间文库测序饱和度)
- 定义: 反映空间文库复杂度的指标,用于评估在当前文库复杂度下,继续增加测序数据量能否有效产出新的空间 UMI 分子。
- 计算方法: Spatial Barcode Saturation = 1 - (Total Spatial UMIs / Valid Spatial Reads)。
- 解读:
- 数值较高:表明测序 reads 中 PCR 重复占比较高,文库接近饱和,继续加深测序的边际收益有限。
- 数值较低:表明文库复杂度仍较高,若空间 UMI 数量不足,可通过增加测序深度来有效提升空间信息量。
Valid Spatial UMIs(有效空间 UMI)
- 定义: 具有有效空间 Barcode 的 UMI 数量,即能够与芯片文库(HDMI 文库)中的已知空间位置成功匹配的 UMI 数。
- 解读: 该指标反映了能够初步建立空间对应关系的分子规模。
- 比例偏低:常见原因包括空间文库构建不佳,或表达文库中较短的 reads 片段错误混入空间文库。建议联查 Valid Spatial Reads 及 Too Short 等结构质控指标。
Spatial UMIs with Unique Locations(具唯一位置的空间 UMI 比例)
- 定义: 在有效空间 UMI 中,其空间 Barcode 能够明确映射至芯片上唯一物理坐标位置的比例。
- 解读: 空间条码若缺乏唯一性,将导致对应的 UMI 无法被可靠地分配到真实的空间坐标并被过滤。
- 比例偏低:提示空间条码存在较高歧义性,这将严重影响后续细胞中心点的准确估计及空间基因表达的还原。
Accurate Spatial UMIs(准确空间 UMI 比例)
- 定义: 在有效空间 UMI 中,经过空间网格过滤,被判定为无明显聚集伪影的“准确空间 UMI”占比。
- 计算方法: 将整个芯片区域划分为规则网格(bins),统计各 bin 内的 UMI 数量。若某一 bin 的 UMI 密度超过所有 bin 平均值的 20 倍,则判定该区域存在技术伪影或污染并予以排除,剩余 UMI 计为准确空间 UMI。
- 解读: 该指标用于剔除空间端可能存在的局部系统性伪影。
- 比例偏低:通常提示空间信号在物理分布上的异常聚集,需结合 Accurate Spatial UMI Bins 及空间距离分布图进行综合排查。
Accurate Spatial UMI Bins(准确空间网格比例)
- 定义: 将芯片划分为网格(bins)后,通过伪影检测判定为“准确(非伪影)”的 bins 在所有 bins 中的占比。
- 计算方法: Accurate Spatial UMI Bins = Accurate Bins Counts / Total Bins Counts。
- 解读: 该指标从空间分布的宏观尺度衡量空间信号是否均匀、可靠。
- 比例偏低:明确指向空间信号在物理分布上的异常聚集,反映可能存在较严重的局部污染或定位误差。
Cell-Identified Spatial UMIs(细胞相关空间 UMI 占比)
- 定义: 在准确的空间 UMI 中,其携带的细胞 Barcode 同时在转录组(RNA)文库中被成功判定为“有效细胞”的 UMI 占比。
- 解读: 该指标衡量双组学数据中细胞识别与空间定位之间的一致性。
- 比例偏低:提示 RNA 侧的细胞判定阈值可能不合适(过宽或过严)、空间定位信号不稳定,或两组学文库在数据质量及细胞捕获效率上存在显著失配。
Mean Spatial UMIs per Cell(每细胞平均空间 UMI 数)
- 定义: 在所有被判定为有效细胞的 Barcode 中,平均每个细胞所分配到的准确空间 UMI 数量。
- 计算方法: 计算所有具有准确空间 UMI 且被判定为细胞的 Barcode 的空间 UMI 数平均值。
- 解读: 直接反映单细胞层级的空间信息丰富度。该数值过低会导致细胞定位不准,或者部分细胞无法计算出唯一中心。
- 异常排查:可以结合“具唯一中心的细胞数占 RNA 细胞数的比例”(如 Number of Cells with Unique Center 相关指标)来综合判断是否属于空间文库构建异常。
Q30 Bases in Barcode / UMI(空间端 Barcode/UMI 的 Q30)
- 定义: 空间文库的测序 reads 中,空间 Barcode 和 UMI 序列的质量值达到或超过 Q30(即碱基识别错误率 ≤ 0.1%)的碱基所占比例。
- 解读: 高 Q30 比例通常表示空间 Barcode 和 UMI 的读取准确性较高。这不仅有助于提高有效空间 Barcode 的识别率与空间定位的稳定性,还能提升 UMI 分子去重的准确性;当该指标偏低时,将直接导致 Valid Spatial Reads 比例下降,并影响下游空间定量结果的可靠性。
Distance Distribution(中心距离分布图)
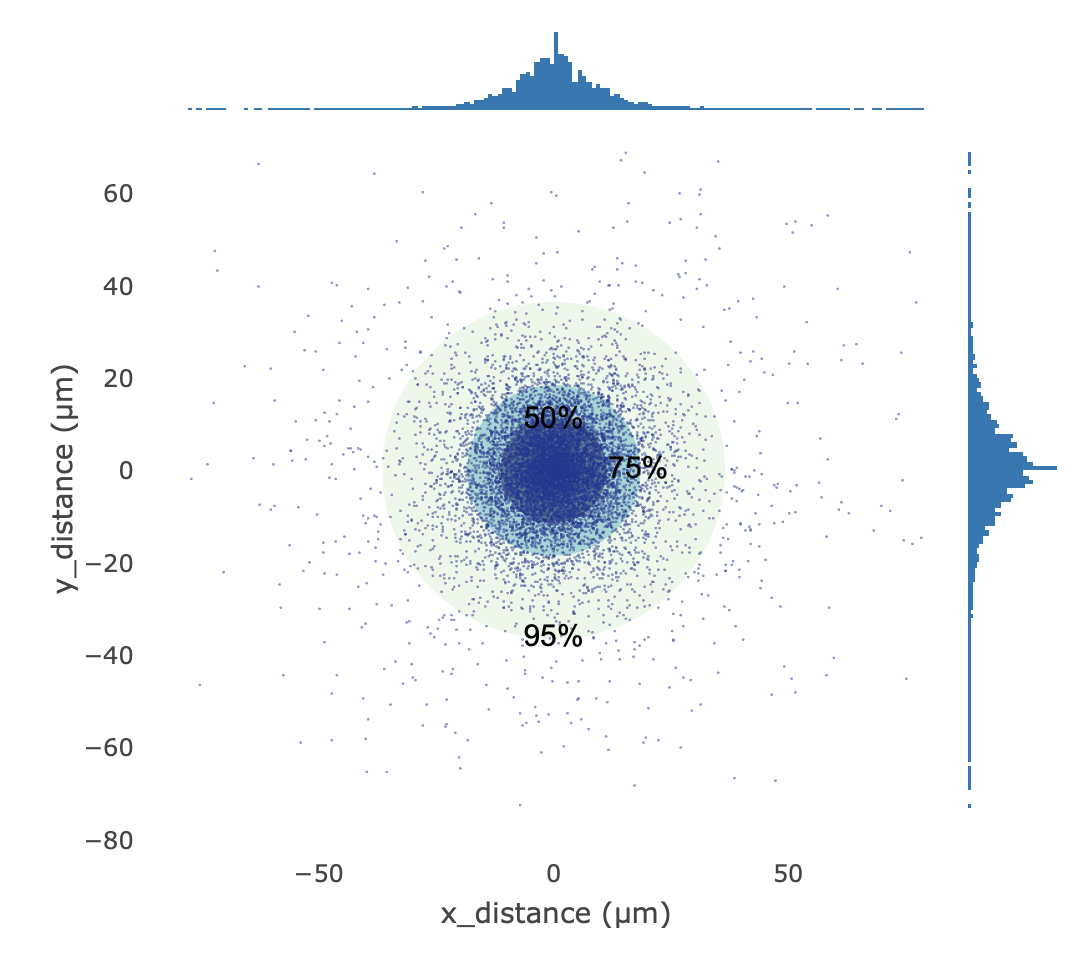
- 定义: 以每个被识别细胞的估计中心点为坐标原点,统计该细胞对应的空间 Barcode / UMI 相对其中心的物理偏移距离分布。
- 计算方法: 随机抽取所有细胞中最多 10,000 个空间 Barcode 坐标点,计算其相对各自细胞中心的 X/Y 轴偏移量,并按特定系数(如 0.265385 µm/pixel)将像素转换为实际物理距离。图中使用 50%、75% 和 95% 三个同心概率圈辅助界定数据点的偏移离散度。
- 解读: 用于直观评估细胞中心估计的收敛性与定位稳定性。
- 理想状态:同心圈较小且数据点高度集中于中心,说明空间定位与中心估计极其稳定。
- 异常状态:若点云显著拉长或整体偏移偏离原点,提示组织切片与芯片定位可能存在整体错位,或细胞中心点估计算法因信号分布异常而产生了系统性偏差。
Sequencing Depth and Saturation Analysis(测序深度与饱和度分析)
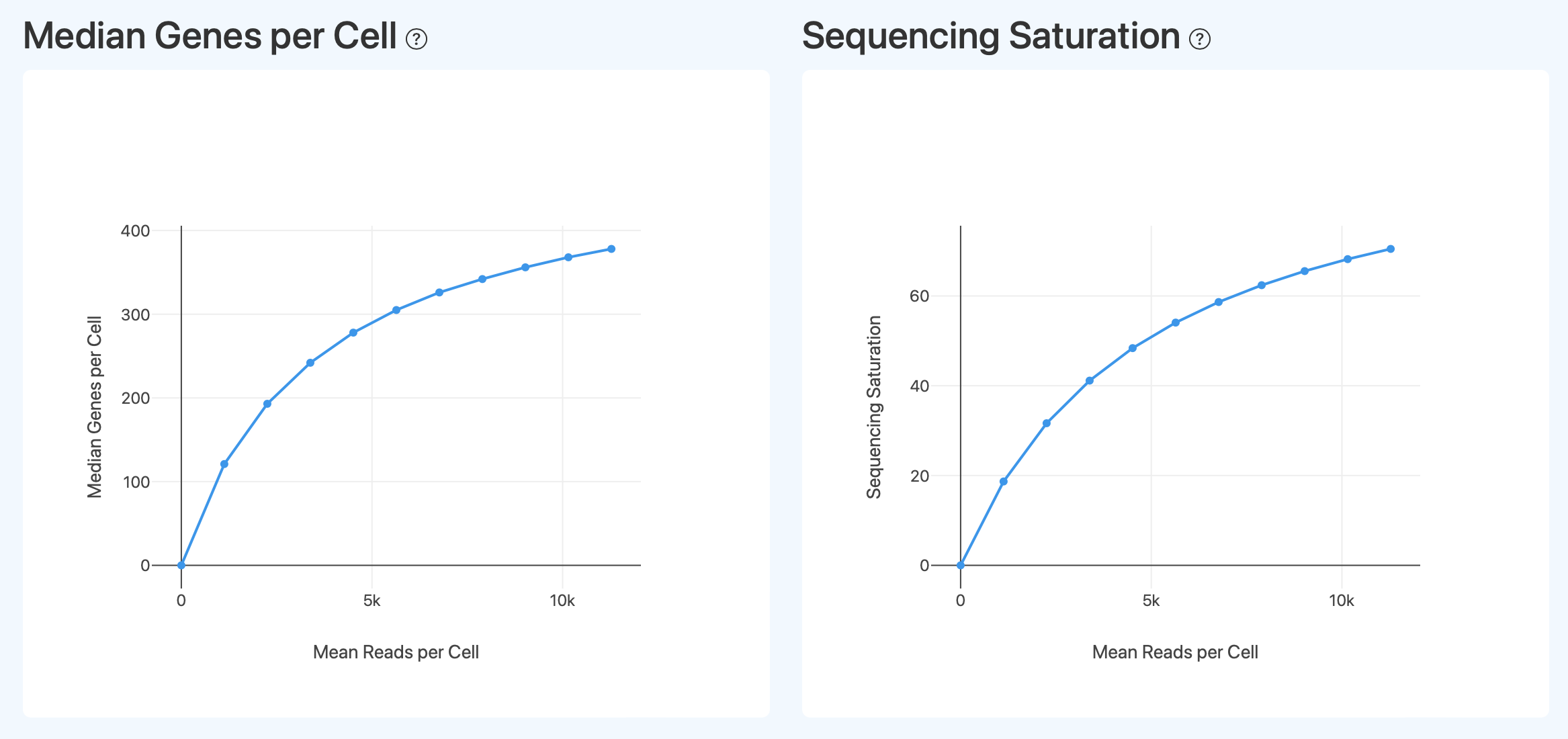
Median Genes per Cell (测序深度与基因检测能力)
- 定义: 基于对 RNA 测序数据进行下采样处理,展示在不同 Mean Reads per Cell 条件下,单细胞 Median Genes per Cell 的变化关系。横轴表示每个细胞的平均测序 reads 数,纵轴表示对应条件下检测到的基因数中位数。
- 解读: 该图用于评估测序深度对单细胞转录组基因检测能力的影响及其饱和趋势。一般而言,随着每个细胞分配的测序 reads 增加,能够检测到的基因数随之上升;当曲线逐渐趋于平缓时,说明新增测序 reads 对基因检测数量的增益开始减弱。该趋势可用于判断当前测序深度是否已接近文库复杂度上限,并为是否需要进一步加深测序提供参考。
Sequencing Saturation(测序深度与重复率关系)
- 定义: 基于对 RNA 测序数据进行下采样处理,展示在不同测序深度条件下,Sequencing Saturation 随测序数据投入变化的趋势。横轴表示下采样后对应的表达信息量水平(以单细胞层面的基因检测情况为参照),纵轴表示测序饱和度,用于反映重复测序分子的比例。
- 解读: 该图用于评估测序深度与文库复杂度之间的关系。随着测序深度增加,测序饱和度通常逐步升高,表示新增 reads 中重复分子的比例增加;当曲线趋于平缓时,说明进一步增加测序深度对获取新的转录分子信息的边际收益有限。该趋势可与基因数或 UMI 数的下采样曲线结合解读,用于综合判断当前测序深度是否已接近文库复杂度上限。
Barcode Rank Plots(Barcode 排序诊断图)
该类图用于直观评估细胞判定边界是否合理,类似于“barcode rank plot”。
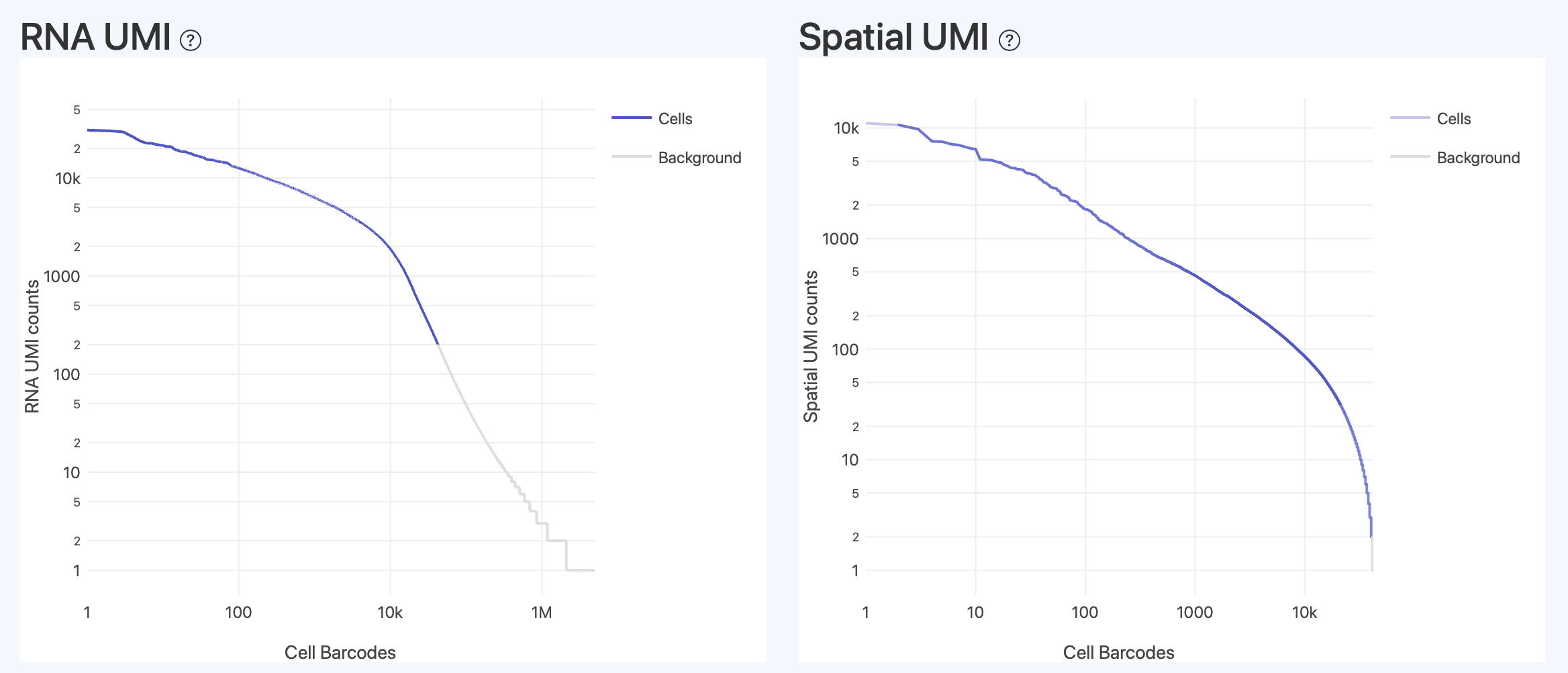
RNA UMI(RNA Barcode UMI 排序)
- 定义: 该图展示所有 Barcode 在 RNA 文库分析流程中用于细胞识别的计数指标分布(以 UMI counts 为例),并按该计数从高到低排序。横轴为排序后的 Barcode(排序位置),纵轴为对应的计数水平。不同颜色用于区分被归类为有效细胞(Cells)与背景(Background)的 Barcode 群体。
- 解读: 该图用于直观查看细胞识别过程中,不同类型 Barcode 在计数强度分布上的整体分离情况。
- 分布趋势: 计数强度较高的区间主要由有效细胞相关的 Barcode 构成,而在计数强度较低的区间,背景(Background)Barcode 的占比逐渐增加。
- 细胞曲线: 其在整体分布中的覆盖范围,可用于观察通过筛选后、用于下游分析的有效细胞在全部 Barcode 中所处的位置及规模。
- 整体核查: 理想形态通常呈现“陡降 + 长尾背景”,细胞区域与背景区域分界相对清晰。不同类型 Barcode 在高、低计数强度区间内的分布关系,以及整体曲线形态,可作为细胞识别与筛选结果合理性的直观核查依据。若过渡区过长或细胞/背景严重混杂,提示细胞判定可能不稳,需结合 Fraction Reads in Cells、Median Genes/UMI 等指标综合判断。
Spatial UMI(空间 Barcode UMI 排序,HDMI/Spatial)
- 定义: 该图展示所有 Barcode 在空间文库分析流程中的计数指标分布(以空间端 UMI counts 为例),并按该计数从高到低排序。横轴为排序后的 Barcode(排序位置),纵轴为对应的空间端计数水平。不同颜色用于区分被归类为“具有唯一有效中心(Cells with unique center)”与背景/无效定位的 Barcode 群体。
- 解读: 该图用于直观查看空间信号在 Barcode 间的分布差异,以及空间定位筛选的效果。
- 分布趋势: 计数强度极高的区间通常代表异常细胞(空间信号过强往往提示空间信号杂乱),正常具有唯一有效中心的细胞多分布于中等强度的平稳区间,而计数强度极低的长尾区间主要为背景或散乱的空间信号。
- 定位细胞曲线: 其在整体分布中的覆盖范围,反映了能够成功估算出唯一中心坐标的细胞群体规模。
- 整体核查: 可用于判断空间端信息是否健康地集中在有效细胞群体中。若空间端 UMI 排序曲线与分布趋势不符,可能提示空间定位异常,需联查 Valid/Accurate Spatial UMIs 与 Unique Locations 等空间质控指标。
Analysis(降维聚类与表达展示)
Analysis 模块用于检查“下游结果是否合理”,但其可信度依赖前述 QC 是否达标。同时,页面右侧提供了交互式的过滤与统计工具,便于用户动态探索数据分布。
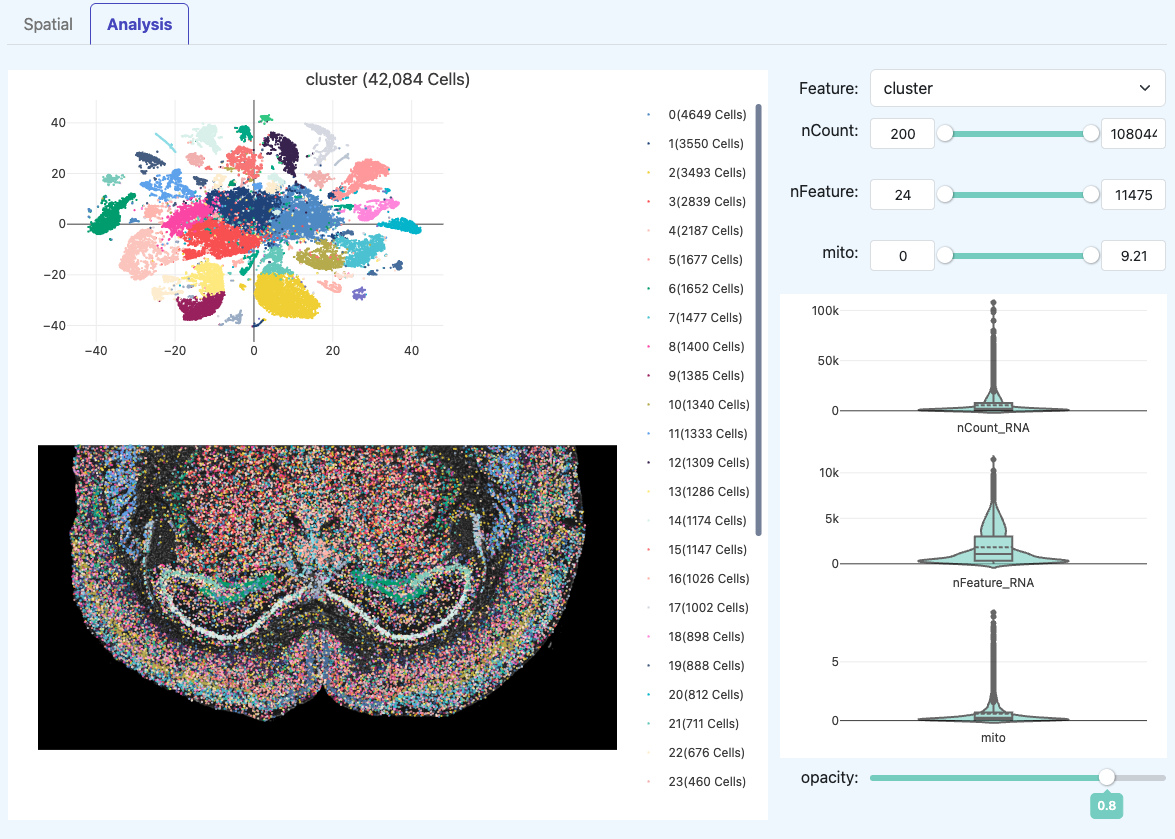
t-SNE 聚类图(RNA)
- 定义: 在有效细胞上进行降维(如 t-SNE)并聚类的二维可视化。
- 解读: 团簇分离清晰通常表示细胞异质性被捕获;若整体糊成一团:可能是数据深度/复杂度不足,也可能是样本本身类型单一;需与 QC 指标一起判断。
组织空间分布图(Spatial)
- 定义: 将聚类标签映射回组织空间坐标,观察不同 cluster 在组织中的位置与形态。
- 解读: 若空间分布与组织结构明显一致,通常支持数据可用;
- 若点位整体偏移或与组织轮廓不一致:优先进行 Adjustment 校准后重跑。
交互式探索工具 (Right Panel)
- 定义: 提供在同一嵌入空间中切换不同特征分布的交互式探索能力。
- 解读: - Feature 下拉菜单:选择不同属性(如
cluster、nCount、nFeature、mito等),切换左侧分布图的着色依据。 - 滑动窗口 (Sliders):调整 nCount、nFeature、mito 显示范围,以及细胞点的透明度 (opacity)。
- 小提琴图 (Violin Plots):动态展示选中细胞群的质量分布特征,辅助判断聚类或空间区域的异常情况。
Marker 展示与差异基因表(DE)
- 定义: 展示各 cluster 的经典标记基因,并按 cluster 输出差异基因表(如 avg_log2FC、p_val_adj 等)。
- 解读: 若 marker 与已知生物学不符:先排除 QC 不达标、细胞混入背景、或参考版本错误等技术问题,再讨论生物学解释。
手动校准(Adjustment)
SeekSpaceTools 质控分析后,图像与细胞位置可能出现不对应情况,需要在报告中手动对齐 DAPI 图像。
操作区域示例:
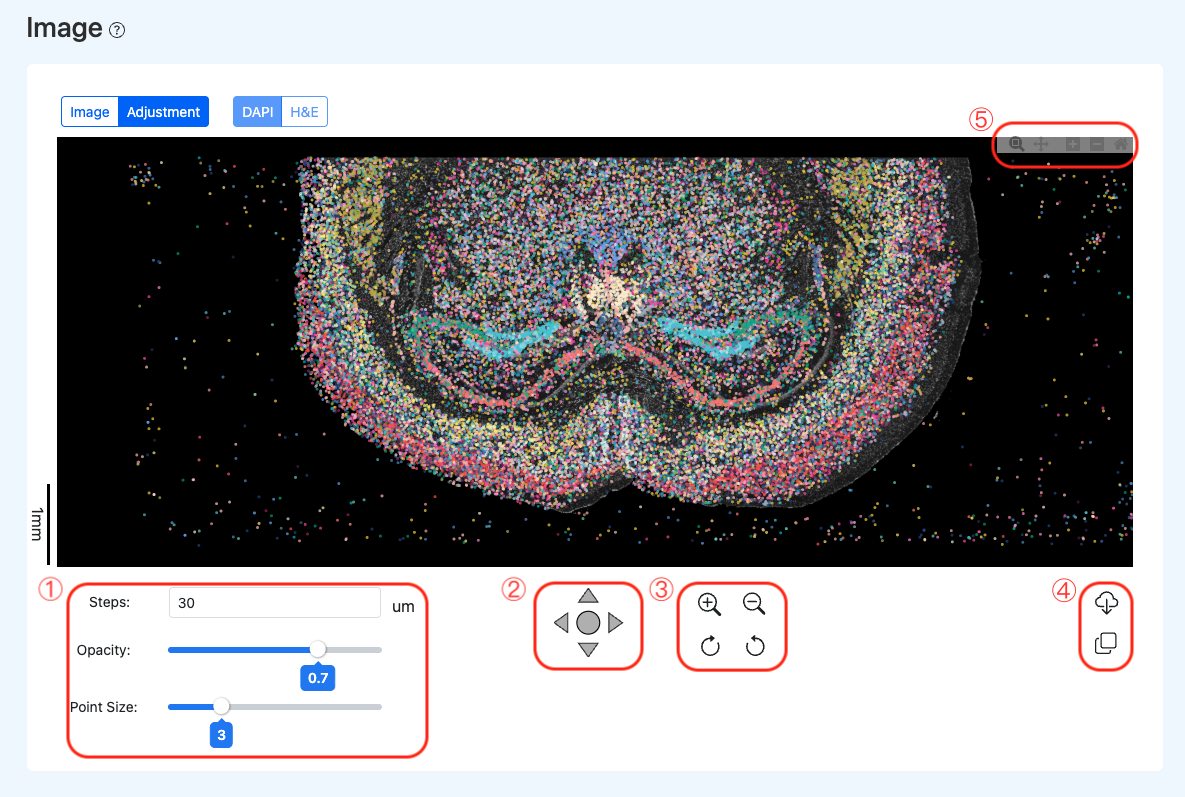
校准工具面板功能说明(对应示例红框):
- 区域 1:可视化参数设置 (Visualization Settings)
- Steps:设置平移步长,用于控制每次点击移动图像时的像素距离,实现精准微调。
- Opacity:调节空间点阵的透明度,便于透过数据点观察底层的组织形态特征。
- Point Size:调整空间数据点的显示大小,以适应不同层级的图像缩放比例。
- 区域 2:平移控制 (Translation Controls):通过方向盘控制底层图像的上下左右平移,实现组织图像与空间数据点阵的精确对齐。
- 区域 3:底图缩放与旋转变换 (Background Scaling & Rotation):提供底层DAPI图像的放大、缩小功能,以及顺/逆时针微调旋转,用于纠正组织切片在成像过程中的微小倾斜与形变(注:此操作仅作用于底图,用于与点阵对齐)。
- 区域 4:校准参数导出 (Export & Reuse)
- Download:下载包含当前对齐参数的校准文件(如 JSON 格式),作为后续重新分析流程的输入。
- Copy:一键复制当前校准参数至剪贴板,方便记录或在批处理中复用。
- 区域 5:全局视图缩放 (Global View Zoom):控制整体视图(包含图像与数据点阵)的放大与缩小,用于深入观察局部区域的对齐细节(注:区别于区域 3 仅对底图的缩放)。
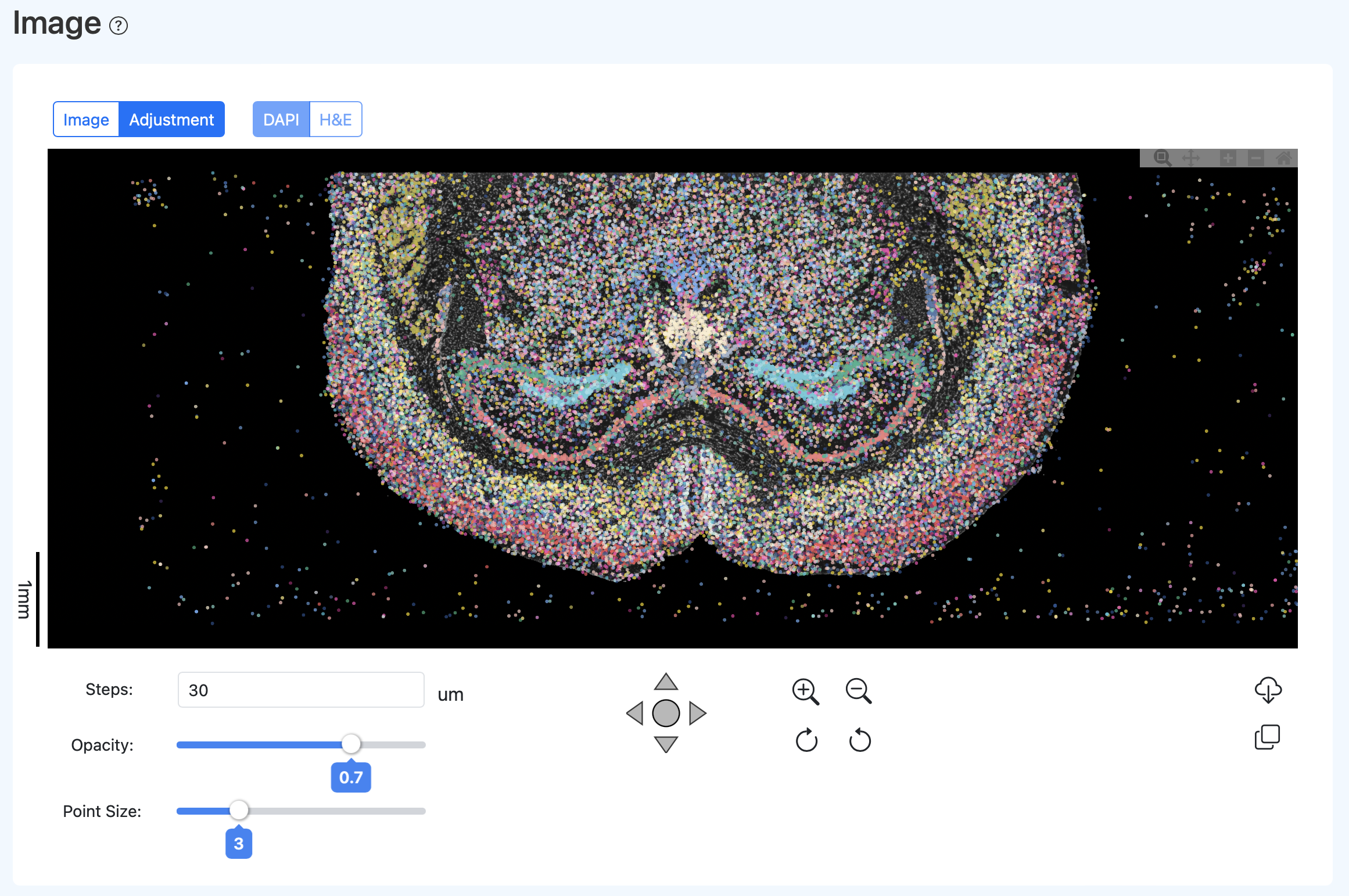
校准建议流程(经验步骤)
- 粗对齐:可先设置 Steps=50um、Opacity=0.2、Point Size=9。让细胞点呈现半透明的组织形态后,移动/旋转/缩放荧光图,使整体轮廓与细胞点云边界大致重合。
- 细对齐:放大全局视图,观察细胞点与细胞核荧光的重合程度,进行小步长微调。目标是多数细胞点与核荧光区域尽可能重合。
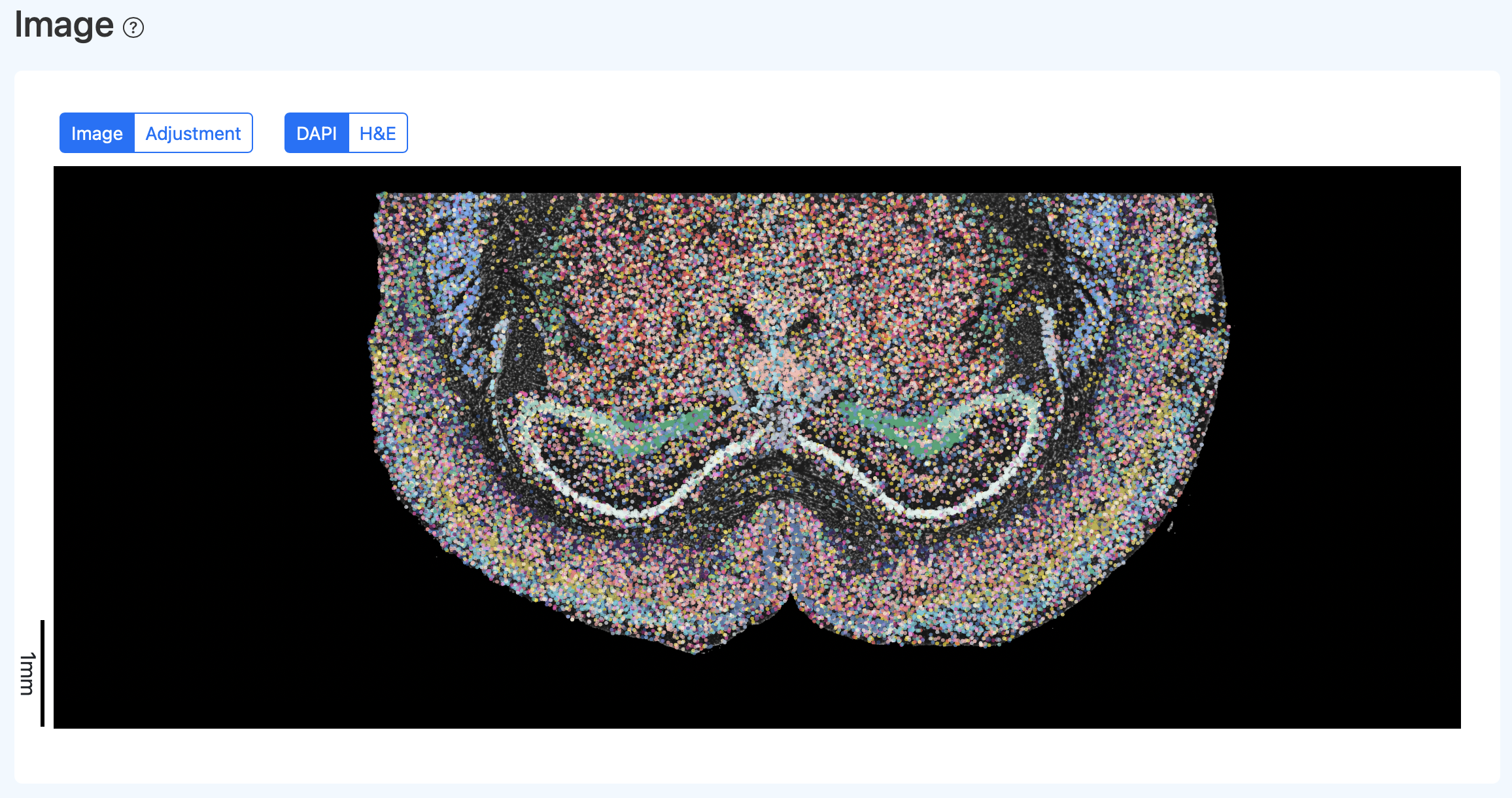
校准后效果示例:
重新生成校准后的报告
- 计算方法:
- 下载调整参数文件;
- 在重新运行 SeekSpaceTools realign 时,将该文件指定给
--alignment_file, 运行图片矫正步骤; - 生成校准后的报告与相关数据。
再次运行后示例:
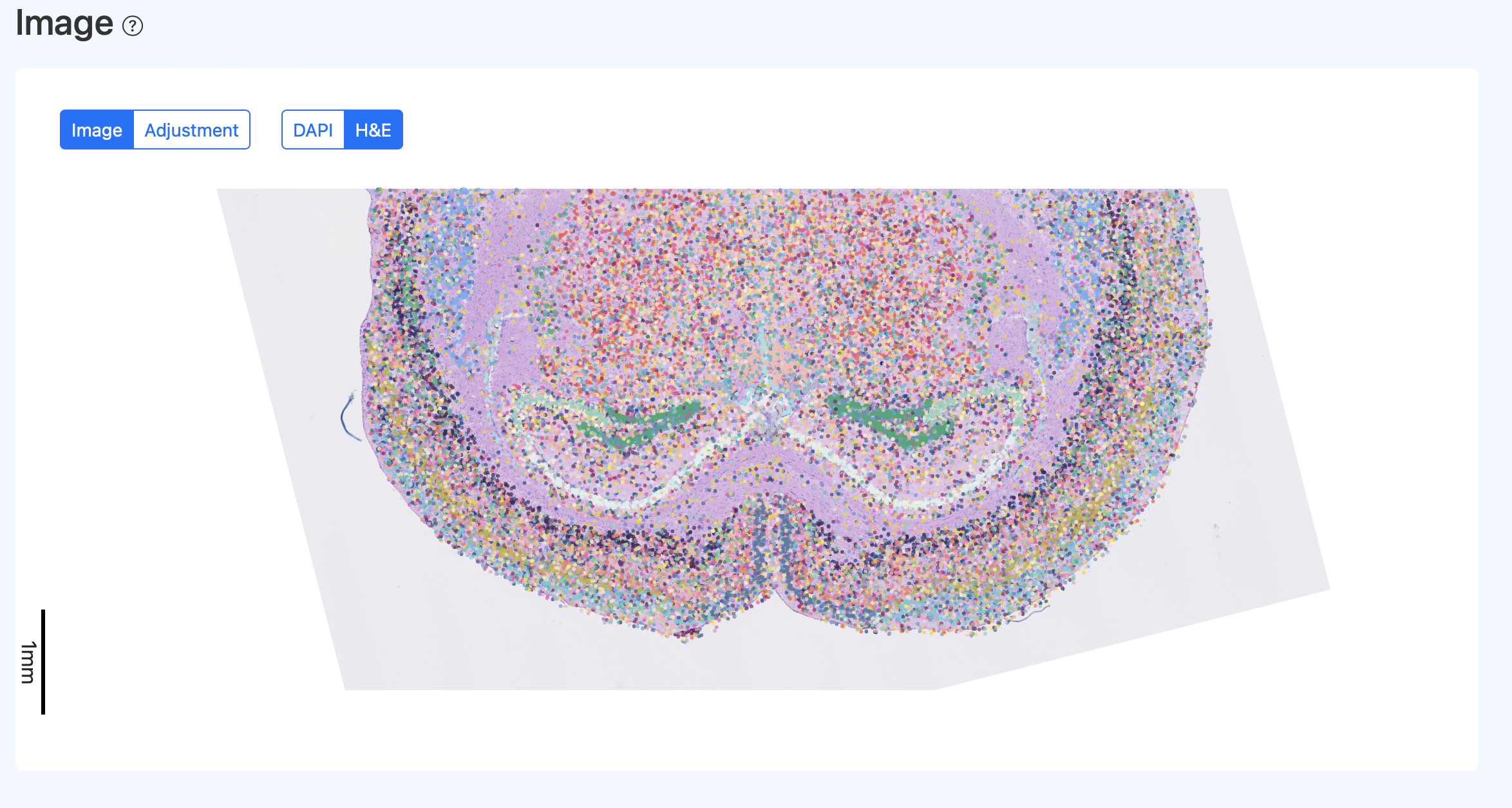
- 解读: 若细胞捕获基于 DAPI 图像而非 HE 图像,通常细胞与 DAPI 的对应更完整;HE 可能存在一定错位,通常属于正常现象。