细胞注释
细胞注释
完成所构建项目的“基础分析”后,可进入“细胞注释”模块。该模块展示细胞注释前后的降维图、cluster差异基因列表及top基因的可视化结果,并可分别对多组聚类结果进行自动注释(SingleR算法)和手动注释。
理论基础
1. 什么是细胞注释?
细胞注释(Cell Annotation)是指根据单细胞基因表达谱,为每个细胞或细胞群赋予其生物学身份(即细胞类型)的过程。这一过程依赖于已知的标记基因(Marker Genes)——那些在特定细胞类型中特异性高表达的基因。
2. 细胞注释的核心原则
NOTE
- 特异性(Specificity):理想的标记基因应仅在目标细胞类型中高表达,在其他类型中不表达或低表达。
- 广度(Coverage):标记基因应在目标细胞类型的大部分细胞中稳定表达。
- 分层性(Hierarchy):细胞身份存在层级关系,如免疫细胞(大群)可进一步分为T细胞、B细胞、巨噬细胞等亚群。注释时应遵循从宏观到微观的原则。
- 多证据支持(Multiple Lines of Evidence):不能仅依赖单个标记基因,应结合多个基因的表达模式、功能富集分析以及参考数据库进行综合判断。
3. 大群注释与亚群注释
- 大群注释(Major-lineage Annotation):识别主要的细胞谱系,如免疫细胞、上皮细胞、基质细胞等。此阶段的目标是建立整个数据集的细胞组成概览。
- 亚群注释(Sub-cluster Annotation):在已确定的大群内部,进一步鉴定更精细的细胞亚型。例如,在T细胞群中区分CD4+ T细胞、CD8+ T细胞、调节性T细胞等。亚群注释是深入研究细胞异质性和功能的关键。
界面说明
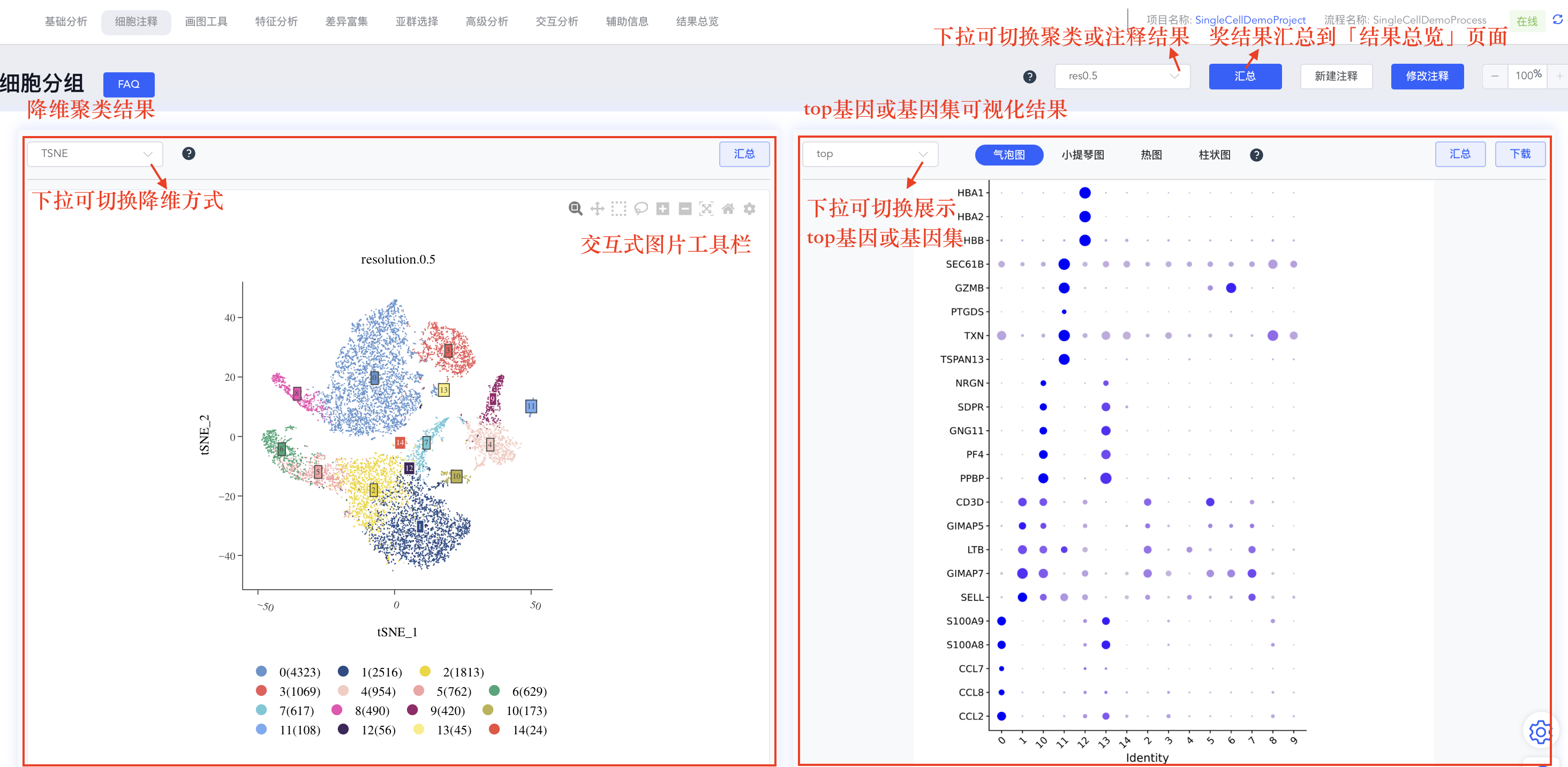
“细胞注释”模块主要包括四部分内容:降维聚类结果、top基因或指定基因集的可视化结果、cluster差异基因列表及差异基因的表达情况。点击选择降维聚类结果,界面中相关结果也会随之发生改变。
- 交互式降维聚类图:降维聚类结果包括UMAP和tSNE两种降维方式,降维聚类图为交互式图片,可使用鼠标更改cluster标签位置和查看cluster的细胞数。
- 差异基因可视化面板:每次更新聚类或注释结果时,均会计算每个cluster的差异基因列表,并对每个cluster的top 5(logFC从大到小排序)基因进行气泡图、小提琴图和热图可视化展示,以及每个样本中每组细胞的占比柱状图。
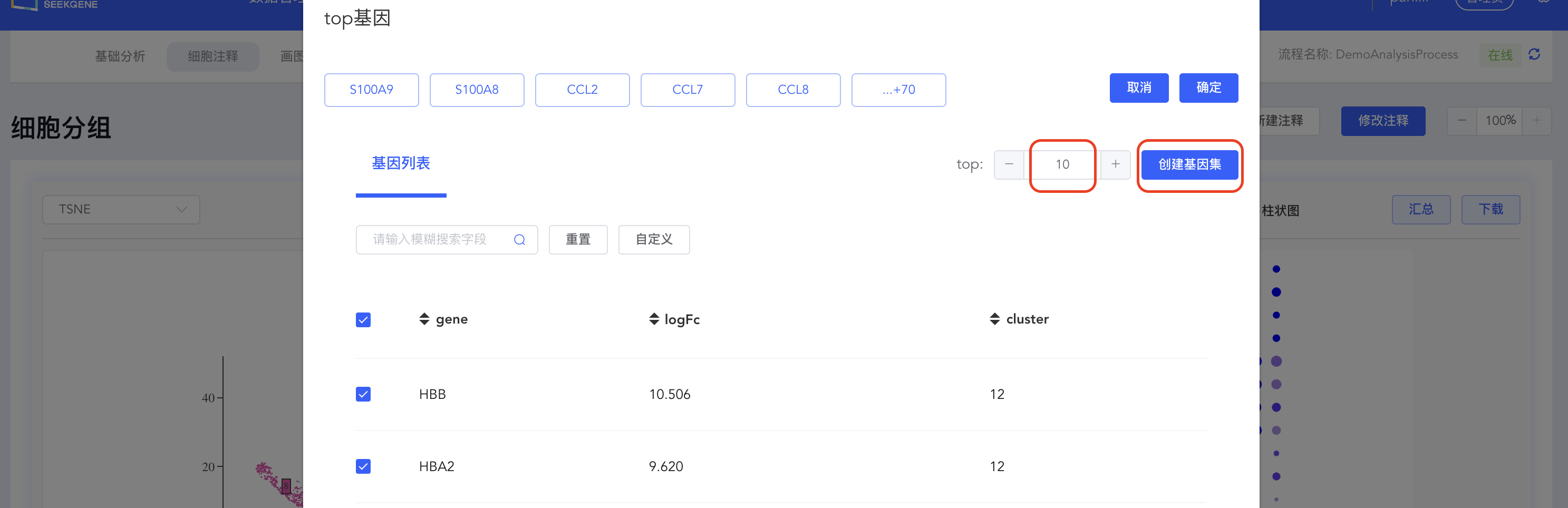
- 自定义top基因集:top基因集可视化结果可自定义修改展示基因。点击【top】按钮,可在弹出窗口中修改用于可视化的基因,勾选或取消勾选对应基因后,点击【确定】即可对所选基因重新绘图;也可指定基因进行可视化展示,点击top下拉菜单中的【基因集】按钮,可从寻因数据库中选择相关基因集,也可点击分析流程基因集后的【+】自定义基因集进行可视化展示。
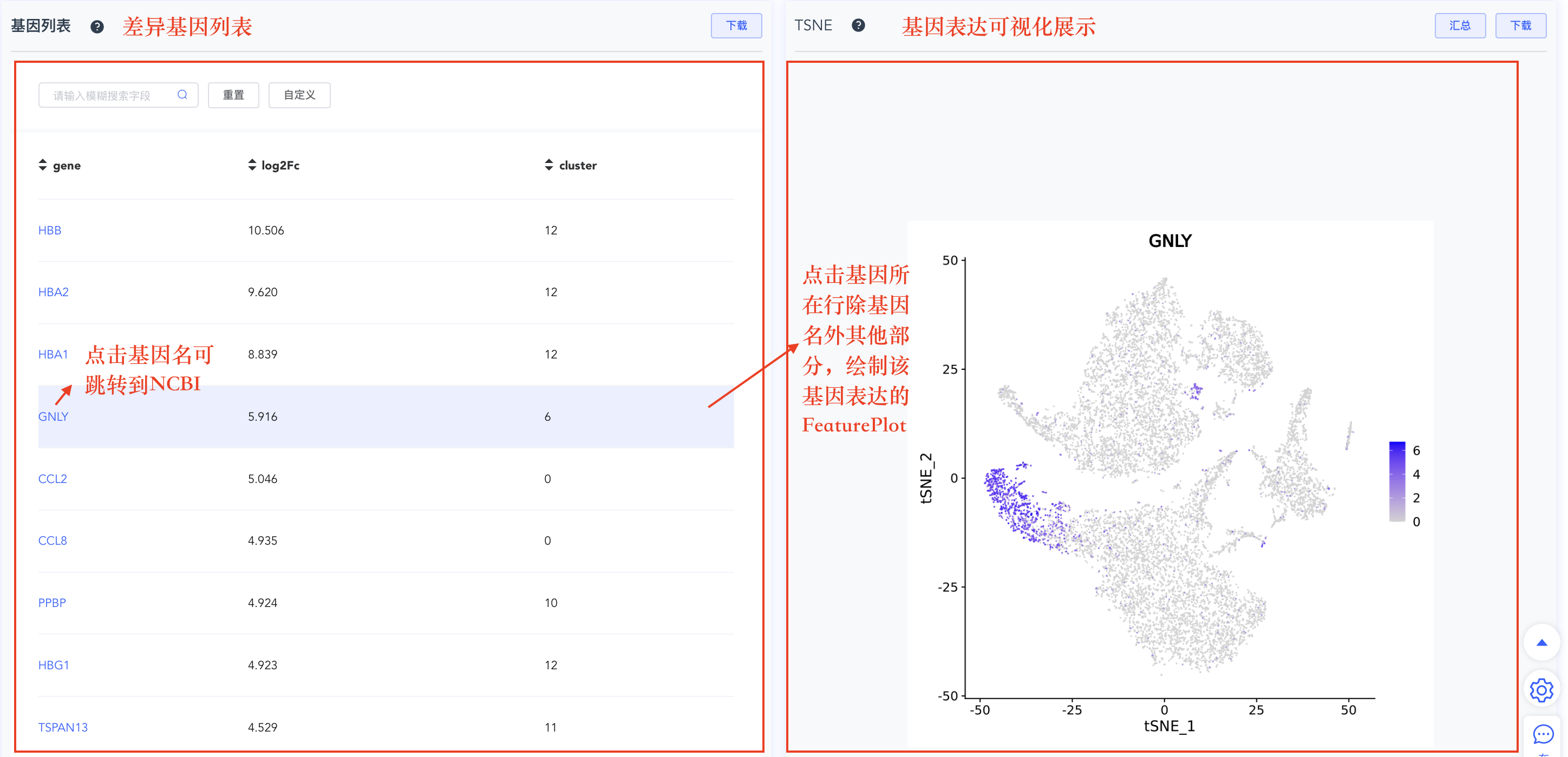
- 差异基因列表:基因列表记录所有基因在某个cluster或分组相对于其他细胞的logFC值。点击基因名称跳转到NCBI网站,点击基因所在行可绘制该基因的FeaturePlot图(位于列表右侧)。该列表默认按logFC从大到小排序,点击表头可自定义排列顺序。
TIP
平台所有的分析结果图,包括降维图、热图等,都可以轻松分享到「结果总览」页面,方便您集中查看、导出和报告。
空间聚类与RNA聚类整合(非必需)
Banksy 与 RNA 聚类的关系
- RNA 聚类仅基于基因表达,擅长区分细胞类型/状态,但可能忽略空间连续性与微环境线索。
- 空间聚类引入邻域依赖与空间梯度,擅长识别组织功能域与边界,但可能牺牲部分表达分辨率。
- Banksy 在表达特征上叠加邻域统计特征(如邻域均值/梯度),通过可调权重进行联合聚类,用于“修正/细化”RNA 聚类边界、合并空间上不连续的伪簇,或揭示被 RNA 聚类忽略的空间功能域。
整合思路(基于云平台工作流)
- 云平台 RNA 聚类基线 → 导入 Banksy CSV → 一致性评估与差异富集
- 在云平台完成标准 RNA 聚类,得到表达层的基线标签。
- 获取高级分析 Banksy 结果 CSV(如
XXX_banksy_colData.csv),通过“上传合并meta”并入流程 meta(须包含barcode,列名避免与流程内置字段冲突)。 - 在平台上对比 RNA 基线与 Banksy 簇/空间域的一致性(NMI/ARI、空间连通性/断裂率、域内表达同质性),并以 Banksy 或一致性优化后的标签为分组做差异表达、富集(GO/KEGG/通路)与空间可视化。
TIP
(为何与如何整合):
- 基于“自身表达 + 空间微环境表达”联合建模:表达决定“它是什么”,空间决定“它在哪里、它做什么”。
- 能识别具有特定空间位置的细胞亚型:揭示仅在特定区域出现的功能性细分亚群。
- 利用邻居信息作为佐证提高置信度:邻域一致的表达模式可帮助合并伪簇、削弱噪声。
- 聚类结果更自然地对齐空间区域:更好地对应组织学结构与形态学边界。
Banksy 并不是替代传统单细胞注释的方法,而是一个强大的增强工具:在既有的 RNA 基线之上注入关键的空间维度,把对细胞身份的理解从“它是什么”推进到“它在哪里、它做什么”的更高层次。
- 云平台 RNA 聚类基线 → 导入 Banksy CSV → 一致性评估与差异富集
关键参数与实践建议(云平台参数面板)
- algo:聚类方法,支持
leiden、louvain、kmeans、mclust。 - 分辨率/簇数:
leiden/louvain用resolution(越大簇越多,结合空间连通性与 marker 解释性微调)。kmeans用kmeans.centers;mclust用mclust.G(均为簇数)。
- lambda:表达与空间位置的权重;
0表示不引入空间,常用0.1–0.3,结构清晰可略升,噪声大/稀疏样本可适度降低避免过度平滑。 - 主成分数量:用于构建特征空间的 PC 数,默认
30,数据规模大或异质性强可适度上调。
- algo:聚类方法,支持
TIP
调参建议:
- 先在 RNA 聚类上确定稳定的表达基线,再在小范围内网格化尝试
lambda与分辨率(或簇数)组合。 - 优先选择兼顾较高 NMI/ARI 与更低空间断裂率的结果;当 NMI 略降但空间域更连续且 marker 更一致时,倾向选择 Banksy 标签。
TIP
实践建议:先固定 RNA 分辨率获得稳定的表达基线,再调节 lambda 与 k_geom 观察空间连通性与 NMI 的变化;当 NMI 下降但空间断裂率显著降低且 marker 呈现更清晰的空间域时,优先选择 Banksy 标签。
- 一致性、解释性与交付
- 一致性:报告 NMI/ARI、空间断裂率、域内表达同质性与空间自相关(如 Moran's I)。
- 可解释性:建立 Banksy 簇与 RNA 类型的映射,核验域内 marker、组织学区域与已知结构的一致性。
- 结果交付:
- 输出“RNA 类型 × 空间域”的交叉表与并排可视化(UMAP 与空间坐标)。
- 基于差异与富集结果,附上显著通路与代表基因的空间热图,阐明空间功能差异。
参考与延伸阅读
Banksy 资料:
注释策略
细胞注释(Cell Annotation)的核心任务是,根据每个细胞或细胞群的基因表达特征,识别出它们在生物学上的真实身份。主流的注释策略分为两大类:
1. 基于标记基因的注释 (Marker-based Annotation)
这是最经典、最依赖专家知识的方法。研究人员根据已知的标记基因(Marker)列表,判断每个细胞cluster的身份。
- 优点:
- 可解释性强:结论直接基于公认的生物学标志物。
- 灵活性高:可根据研究的具体情境,调整和优化Marker列表。
- 能发现新细胞类型:对于无法被现有数据库匹配的未知细胞群,此方法是唯一有效的鉴定手段。
- 缺点:
- 主观性强:高度依赖研究者的先验知识,不同的人可能会有不同的判断。
- 知识局限性:对于研究不充分的组织或物种,可能缺乏可靠的Marker基因。
- 耗时耗力:需要手动检查大量基因的表达模式。
2. 基于参考数据集的注释 (Reference-based Annotation)
该方法将待查询的单细胞数据与一个已经注释好的、高质量的参考图谱(Reference Atlas)进行比对,通过计算相似性来自动分配细胞身份。
- 优点:
- 客观、自动化:减少了人为干预,结果可重复性高。
- 高效快捷:能够快速对大规模数据集给出初步注释。
- 缺点:
- 依赖参考图谱质量:如果参考图谱本身不准确或不完整,会导致错误的注释。
- 无法识别新类型:只能识别出参考图谱中已存在的细胞类型。
IMPORTANT
在实践中,最佳策略是将两者结合。先使用自动化注释(如平台内置的SingleR)获得一个快速、客观的基线结果,然后基于已知的Marker基因进行手动验证和精修。这既保证了效率,又确保了结果的准确性。
实操指南
自动注释
“细胞注释”模块可进行自动注释和手动注释。在完成“基础分析”后,会基于最小分辨率,使用SingleR软件及其参考集(人:HumanPrimaryCellAtlasData_main,小鼠和大鼠:ref_Mouse_all)进行自动注释,自动注释标签为CellAnnotation。
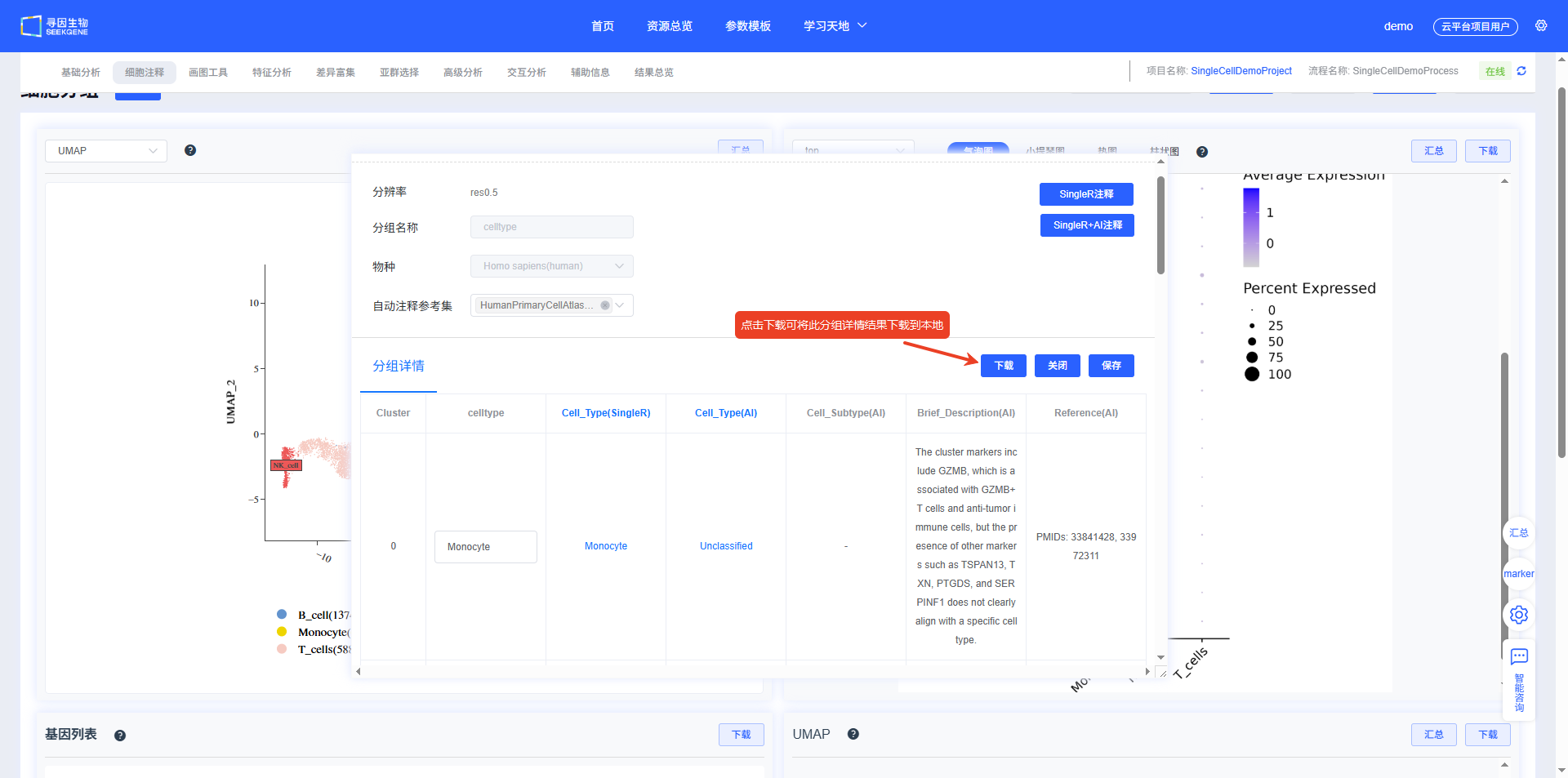
- 首先下拉选择自动注释要使用的分辨率,然后点击【新建注释】,弹出添加分组弹窗,该弹窗可随意拖动位置(鼠标放置在可拖拽区域)和大小(鼠标放置在弹窗右下角)。
- 填写“分组名称”,并选择“自动注释参考集”,最后点击【自动注释】,自动刷新分析进度。
- 分析进度完成后,对应注释结果自动填充到分组详情表中。同时,降维图、top基因可视化结果、基因列表和基因可视化图同步更新。注释完成后点击弹窗底部的【关闭】关闭弹窗。
- 点击同一分辨率下新增分组名称的注释标签,可进行结果切换。
- 当点击【Single+AI注释】时,会得到Single和AI的注释结果,以及参考的marker描述和文献。
手动注释
手动注释是保证最终结果准确性的关键环节。当依据cluster间的差异基因或收集到的marker基因表达情况,需要输入或修改cluster注释结果时,可进行如下手动注释操作:
- 下拉选择手动注释要使用的分辨率,然后点击【新建注释】,弹出添加分组弹窗,该弹窗可随意拖动位置(鼠标放置在可拖拽区域)和大小(鼠标放置在弹窗右下角)。
- 填写“分组名称”,在分组详情中输入每个cluster对应的细胞类型。
- 每个cluster对应的细胞类型输入完成后,点击弹窗底部的【保存】,出现分析进度条,完成后点击【关闭】按钮关闭弹窗。
- 点击同一分辨率下新增分组名称的注释标签,可进行结果切换。
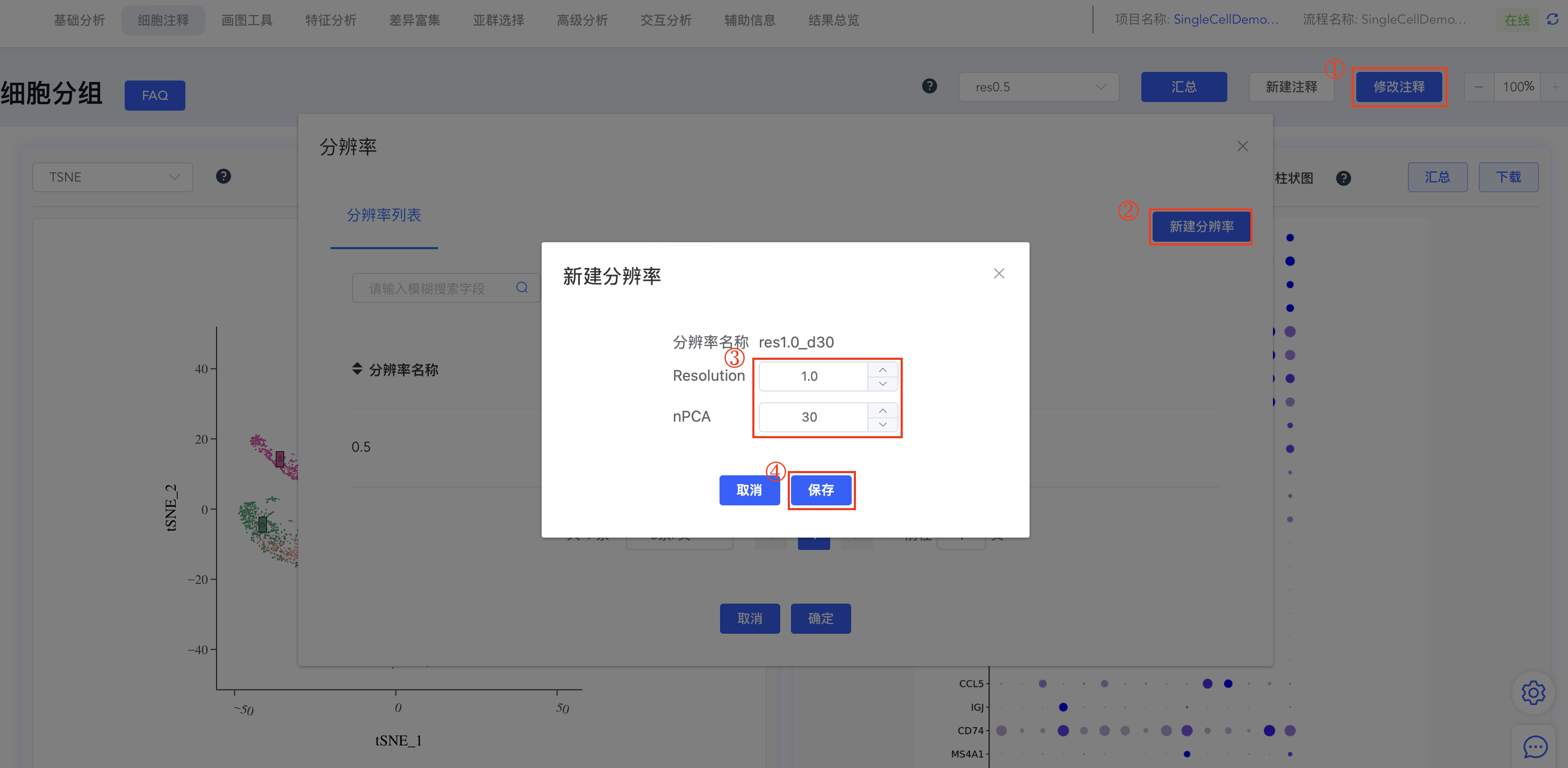
- 新建分辨率:
- 点击细胞分组中的【修改注释】弹出分辨率窗口,点击分辨率列表后的【+】弹出新增分辨率窗口,输入分辨率(Resolution)和PC数量(nPCA)后,点击【保存】,即可基于新的分辨率和PC数量重新进行聚类分析。完成后,细胞分组下拉框和分辨率列表均新增对应结果,点击细胞分组可进行结果切换。
- 修改注释结果:
- 点击细胞分组中的【修改注释】弹出分辨率窗口,点击分辨率对应的【编辑】可修改注释结果。如果该分辨率未做任何注释,则弹出“暂无分组”的提示信息;点击【删除】即可删除该分辨率及对应的细胞注释结果。
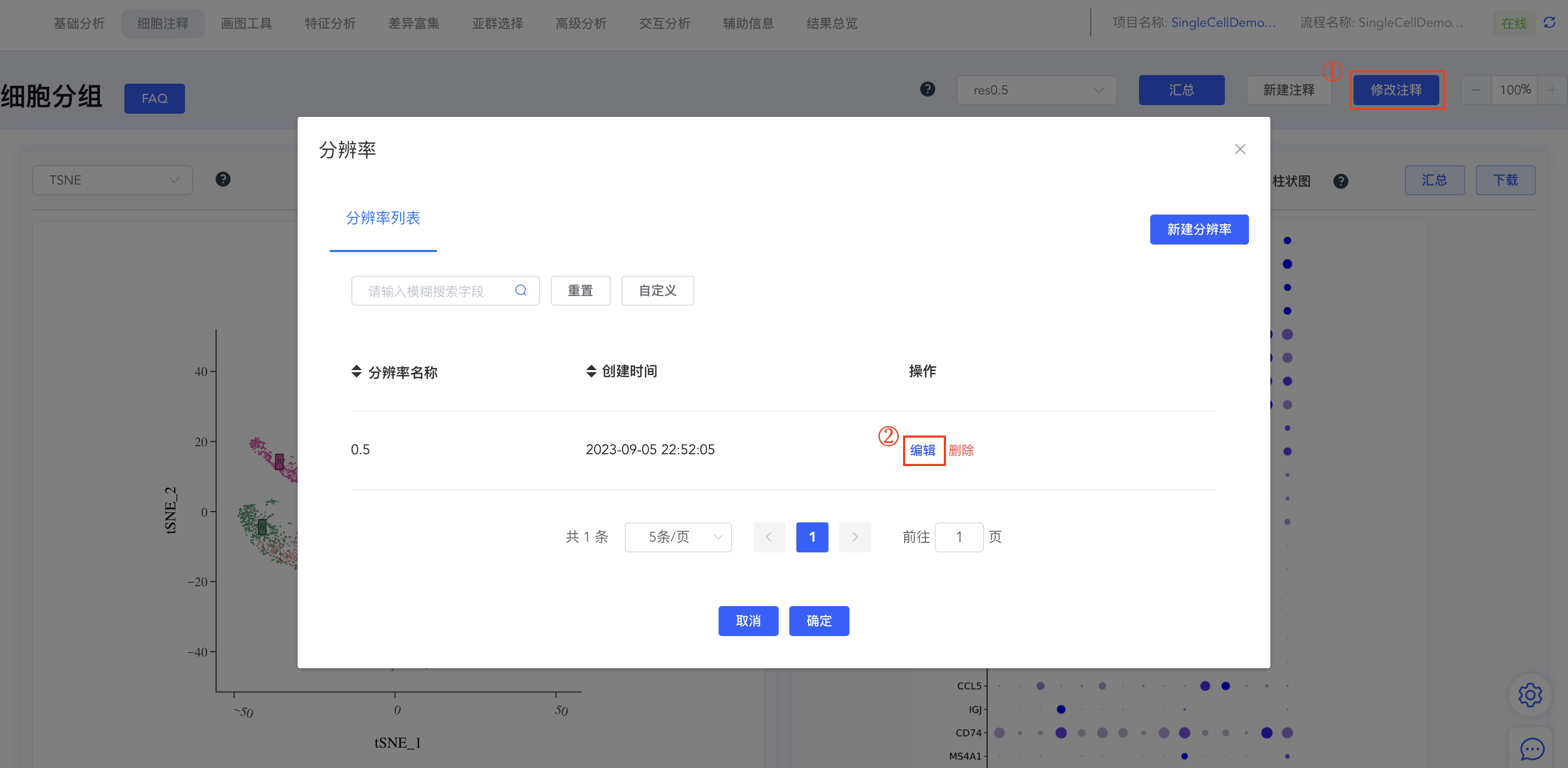
- 保存注释结果:
- 点击【编辑】后弹出编辑分组弹窗,分组详情中包括此分辨率下所有cluster的标注结果,可进行相应编辑,修改完成后点击【保存】同步结果。点击【x】可删除该分辨率下的该组注释结果。
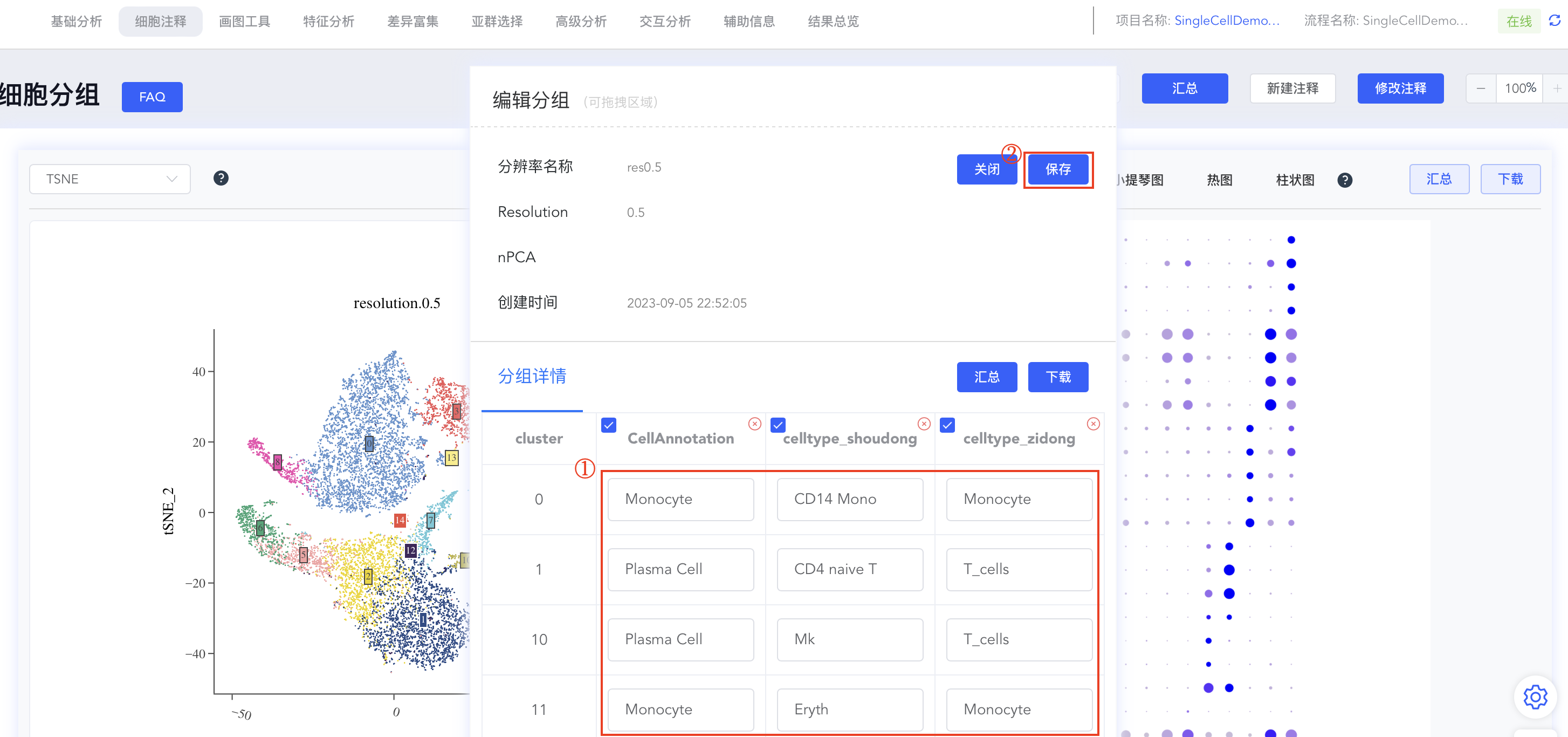
TIP
可针对同一降维聚类结果多次注释并修改注释,比较不同方法和参考集的注释差异。
借助“寻小因”辅助注释
在手动注释过程中,研究者往往需要依赖先验知识和大量文献来确定细胞类型。为了提高注释的效率和准确性,“寻小因”智能助手为您提供了强大的支持。点击页面右下角侧栏的【智能咨询】,即可打开“寻小因”智能助手进入细胞注释界面。

什么是“寻小因”?
“寻小因”是平台内置的AI助手,它整合了海量生物学知识库。您可以通过自然语言对话,快速查询细胞标记基因、获取相关文献、解答生物学问题,从而辅助您完成细胞注释。
如何使用“寻小因”?
- 查询标记基因:当您对某个细胞群的身份不确定时,可以向“寻小因”提问。例如,您可以直接问:“耗竭T细胞的marker有哪些?”
- 获取参考信息:“寻小因”会返回相关的标记基因列表,并附上参考文献(PMIDs),为您提供决策依据。
- 验证与注释:根据“寻小因”提供的信息,您可以在平台的“差异基因可视化面板”中检查这些基因的表达情况,以验证您的假设,并完成对细胞群的精准注释。
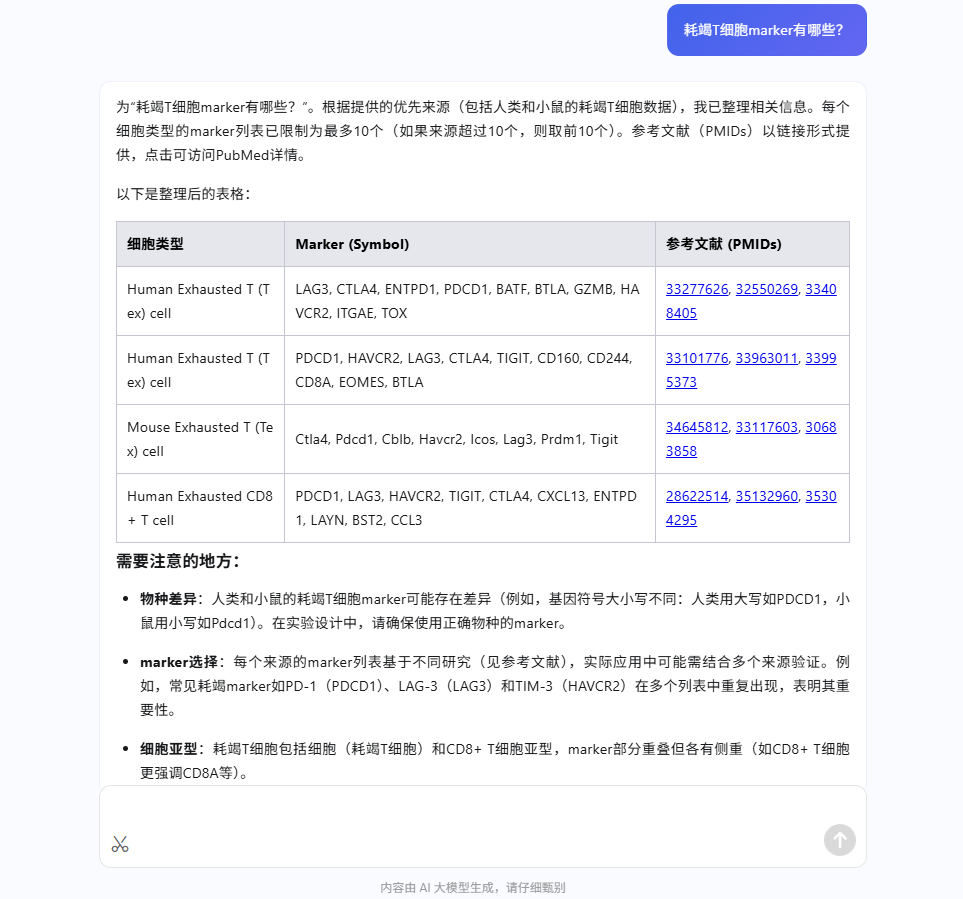
通过结合“寻小因”的智能问答与平台强大的可视化功能,您可以将手动注释的严谨性与AI的便捷性融为一体,显著提升研究效率。
创建top N基因集
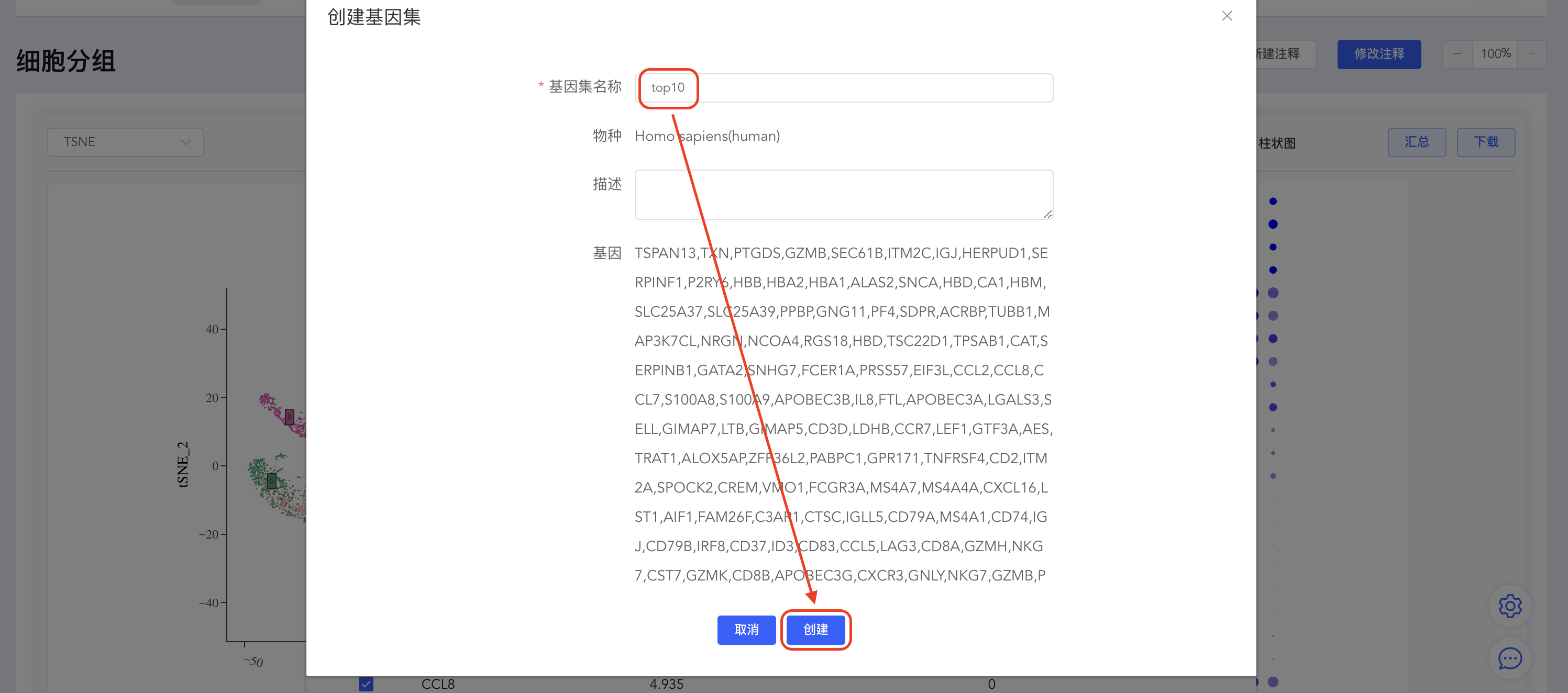
点击【top】按钮打开top基因界面,填写top基因数量(如top10),点击【创建基因集】,填写基因集名称,点击【创建】完成基因集的创建。
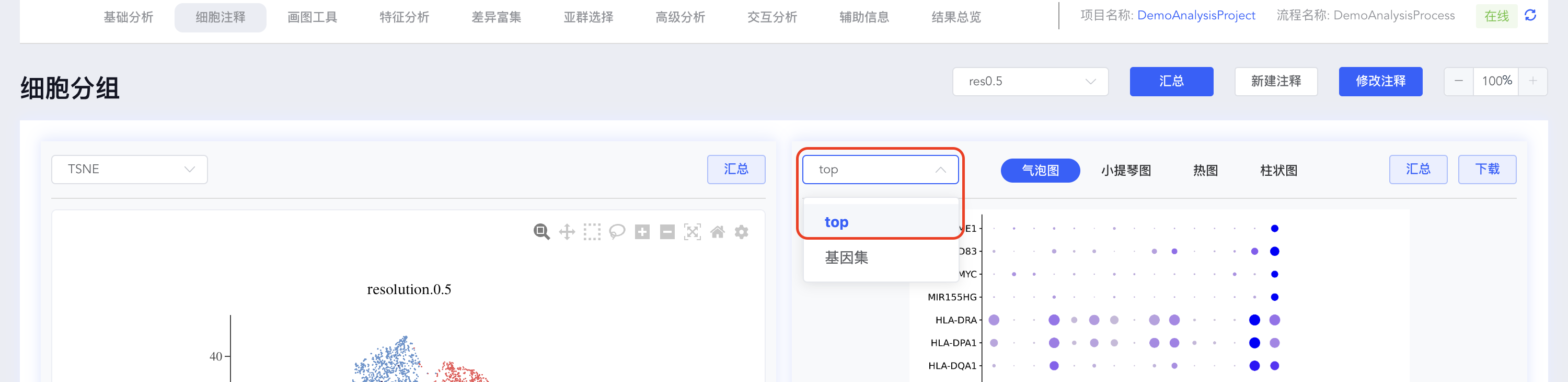
注意事项
当细胞数量过大时,热图会丢失label等信息,因此热图是经过降低数据量(downsample)绘制的,每个cluster或分组最多保留500个细胞;
为加快计算差异基因速度,该模块基因的logFC是基于presto包计算得到的,与FindAllMarkers或FindMarkers结果会存在些许差异,但两种方法计算出的基因呈线性相关;
特殊物种的注释可参考动植物特殊物种的单细胞注释方法讨论
结果解读
一份高质量的细胞注释结果,需要从以下几个方面进行解读和评估:
- 降维图的一致性:在UMAP/tSNE图上,相同类型的细胞是否聚集在一起?不同类型细胞之间是否有清晰的界限?如果界限模糊,可能意味着这些细胞类型在转录组上本身就比较相似,或者需要调整聚类参数。
- Marker基因的特异性:通过热图、小提琴图和FeaturePlot,检查您的关键Marker基因是否在预期的细胞群中特异性高表达。例如,
CD4基因应该只在CD4+ T细胞中亮起。 - 细胞比例的合理性:注释出的各类细胞的比例是否符合样本的生物学背景?例如,在健康人的外周血中,淋巴细胞(T、B、NK细胞)通常是主要成分。
- 生物学故事的完整性:最终的注释结果能否讲述一个通顺的生物学故事?例如,在肿瘤样本中,是否同时鉴定出了肿瘤细胞、免疫细胞和基质细胞,这符合肿瘤微环境的基本构成。
应用示例:肿瘤微环境中的T细胞亚群注释
- 背景:在一份肺癌组织的单细胞测序数据中,我们已经完成了大群注释,识别出了T细胞群体。
- 目标:深入分析T细胞的异质性,寻找可能影响免疫治疗效果的特定T细胞亚群。
- 方法:对T细胞群体进行亚群注释,结合经典Marker(
CD4,CD8A,FOXP3,PDCD1(PD-1),CTLA4)进行鉴定。 - 发现与解读:我们识别出了多个T细胞亚群,包括一个同时高表达
PDCD1和CTLA4的CD8+ T细胞亚群。这一发现揭示了肿瘤内存在一群功能耗竭的T细胞,这可能是导致免疫治疗效果不佳的原因之一,为后续研究提供了方向。