基础分析
时长: 6 分钟
字数: 1.5k 字
更新: 2025-09-15
阅读: 0 次
“基础分析”是分析流程的初始模块,可进行基因、线粒体过滤、去批次整合聚类,保留质量合格的细胞用于后续分析。分析流程创建及过滤整合聚类流程如下:
初始阶段
【新建流程】创建单细胞分析流程。项目开始一般选择样本进行大群分析,后续可选择注释好的细胞类型进行亚群分析。
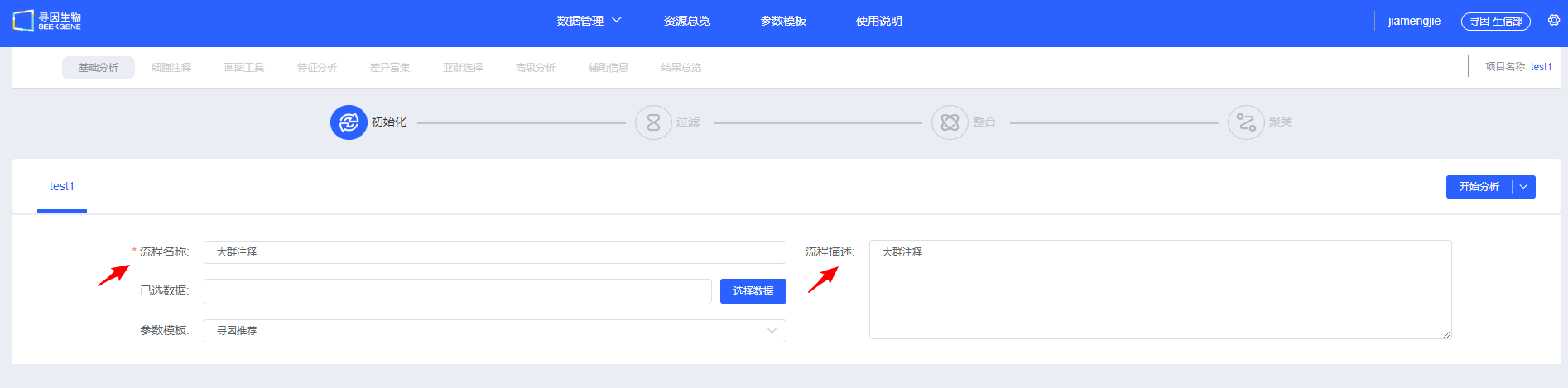
填写“流程名称”及“流程描述”,用于后续查找和了解流程信息。
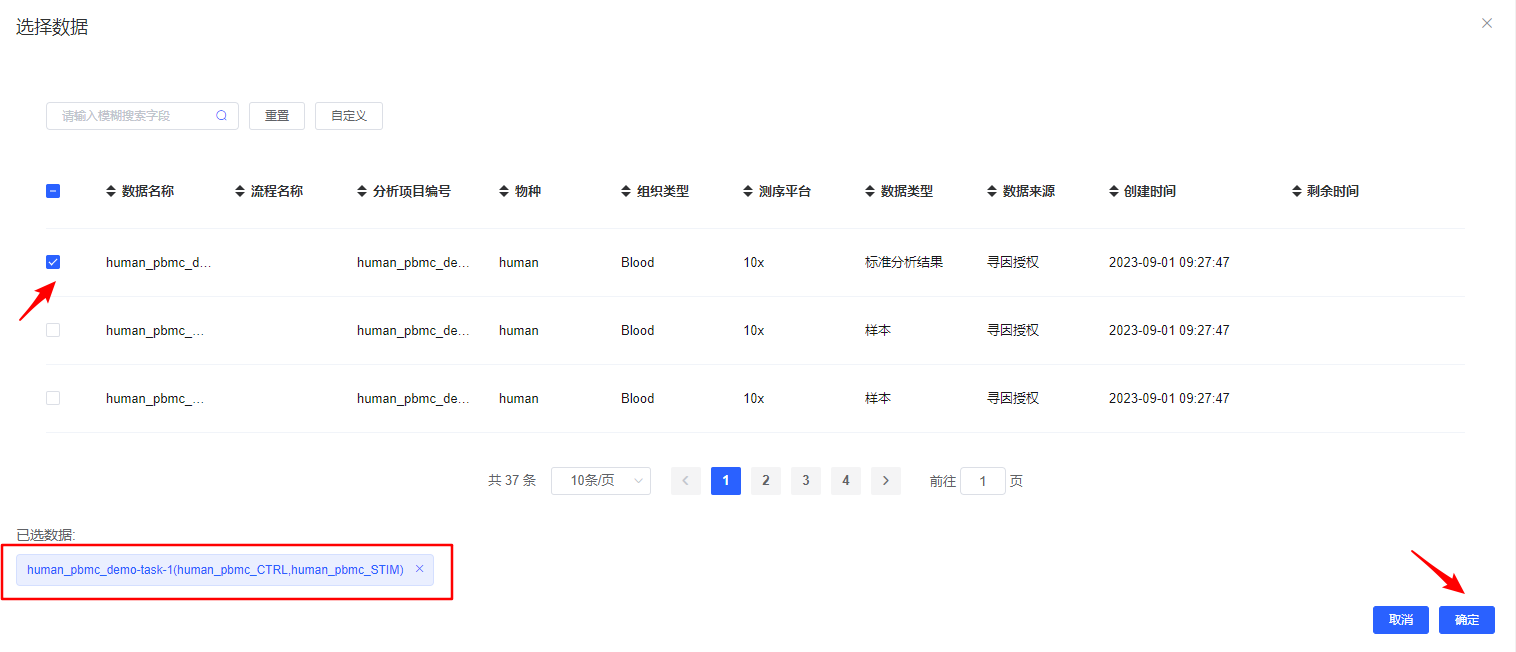
【选择数据】选择待分析的样本,可选择已经整合好的多样本数据,也可选多个单样本进行过滤整合。
NOTE
数据详细来源可查看《我的数据》。
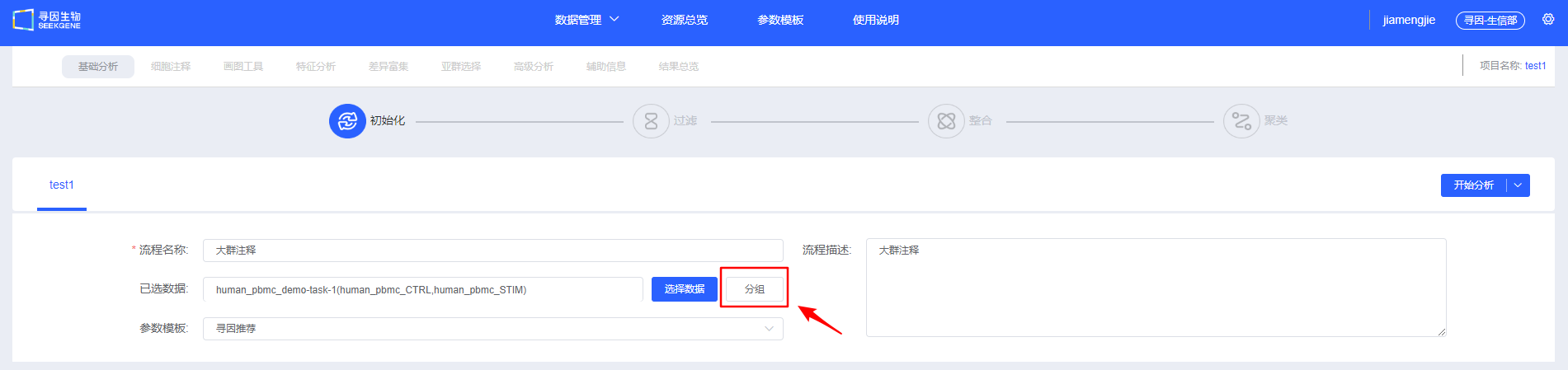
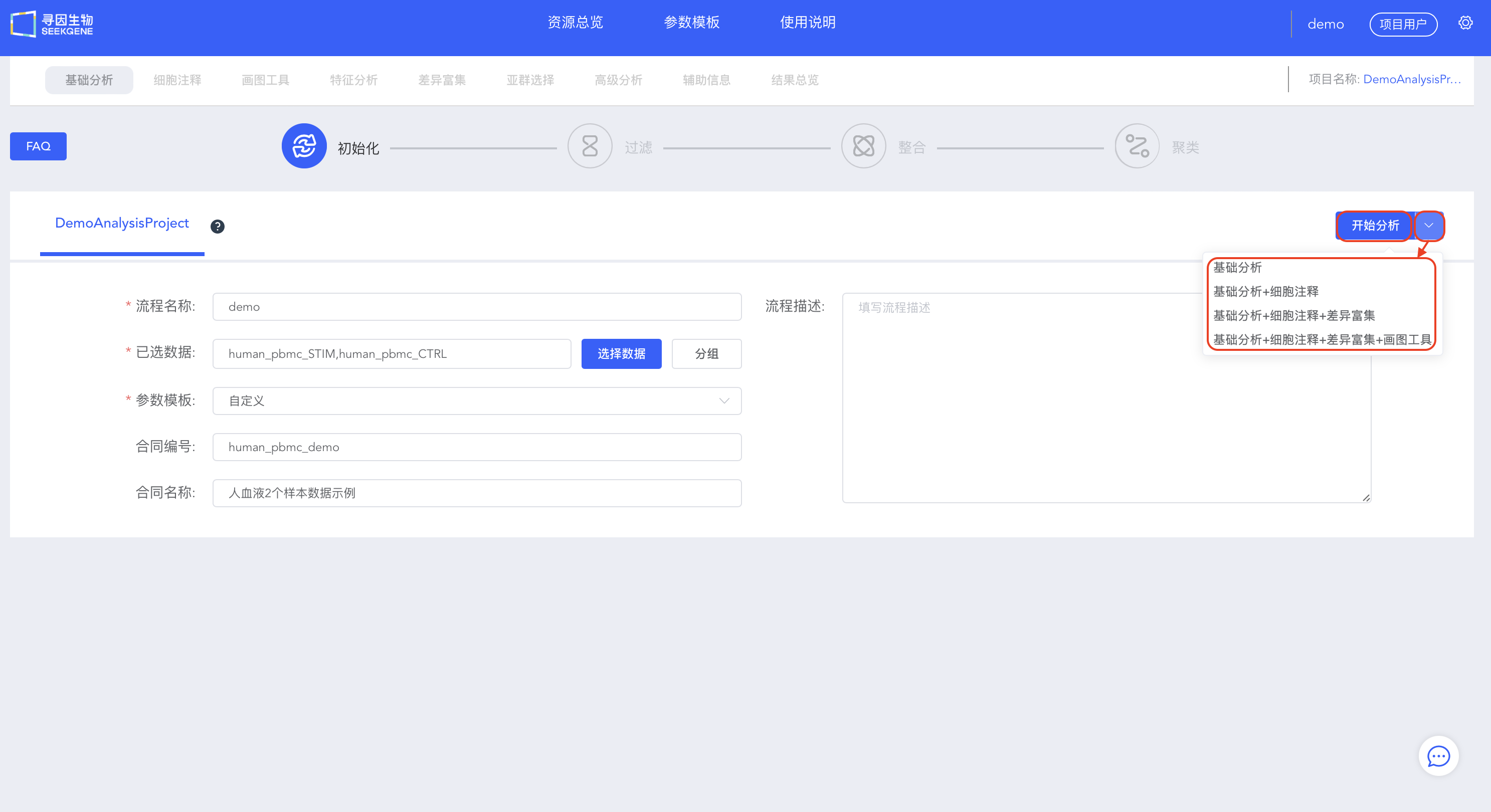
- 填写信息,选择待分析的数据,可以选择【开始分析】手动进行过滤整合和聚类,也可以选择【基础分析】、【基础分析+细胞注释】、【基础分析+细胞注释+差异富集】或【基础分析+细胞注释+差异富集+画图工具】快速进行自动分析。
过滤
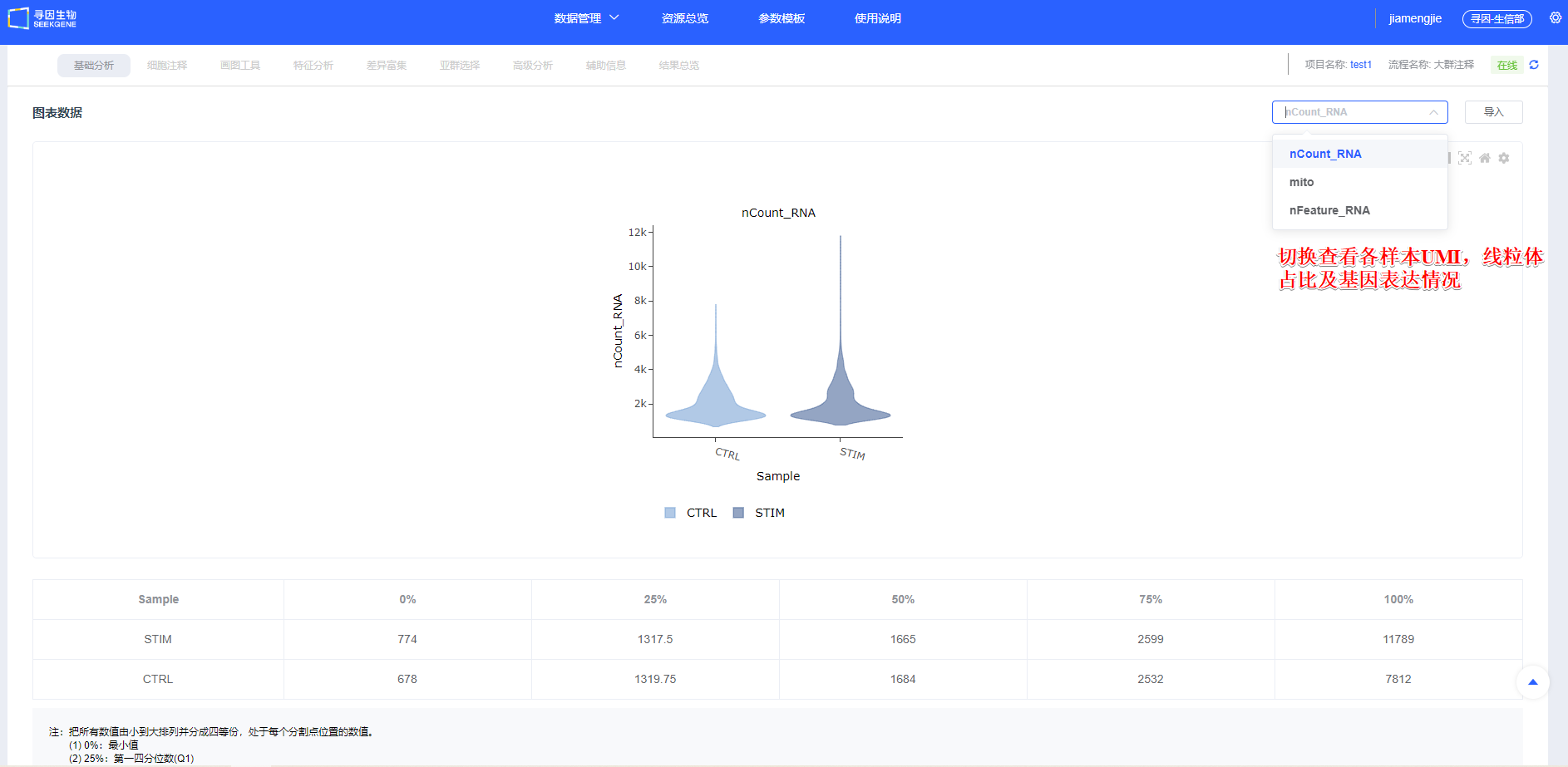
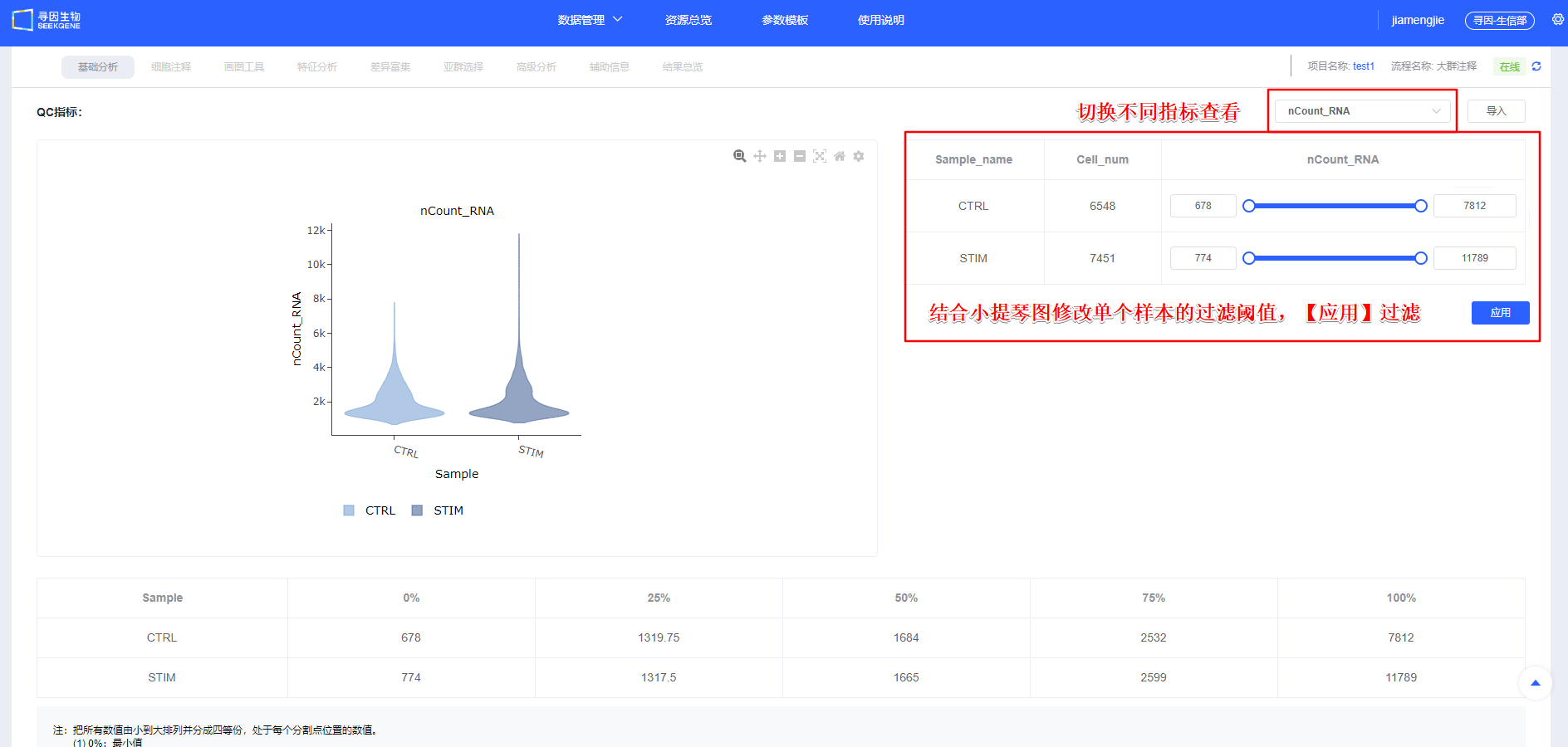
- 【开始分析】后的“图表数据”会统计样本UMI、线粒体占比及基因表达情况,简要展示各样本质量信息。
NOTE
指标释义(scRNA-seq):
- nCount_RNA:每细胞 UMI 总数,反映测序深度与转录本丰度
- nFeature_RNA:每细胞检出的基因数,反映表达复杂度
- 线粒体比例(mito):线粒体基因 UMI 占比,偏高常与凋亡/损伤相关(高代谢组织可适度放宽)
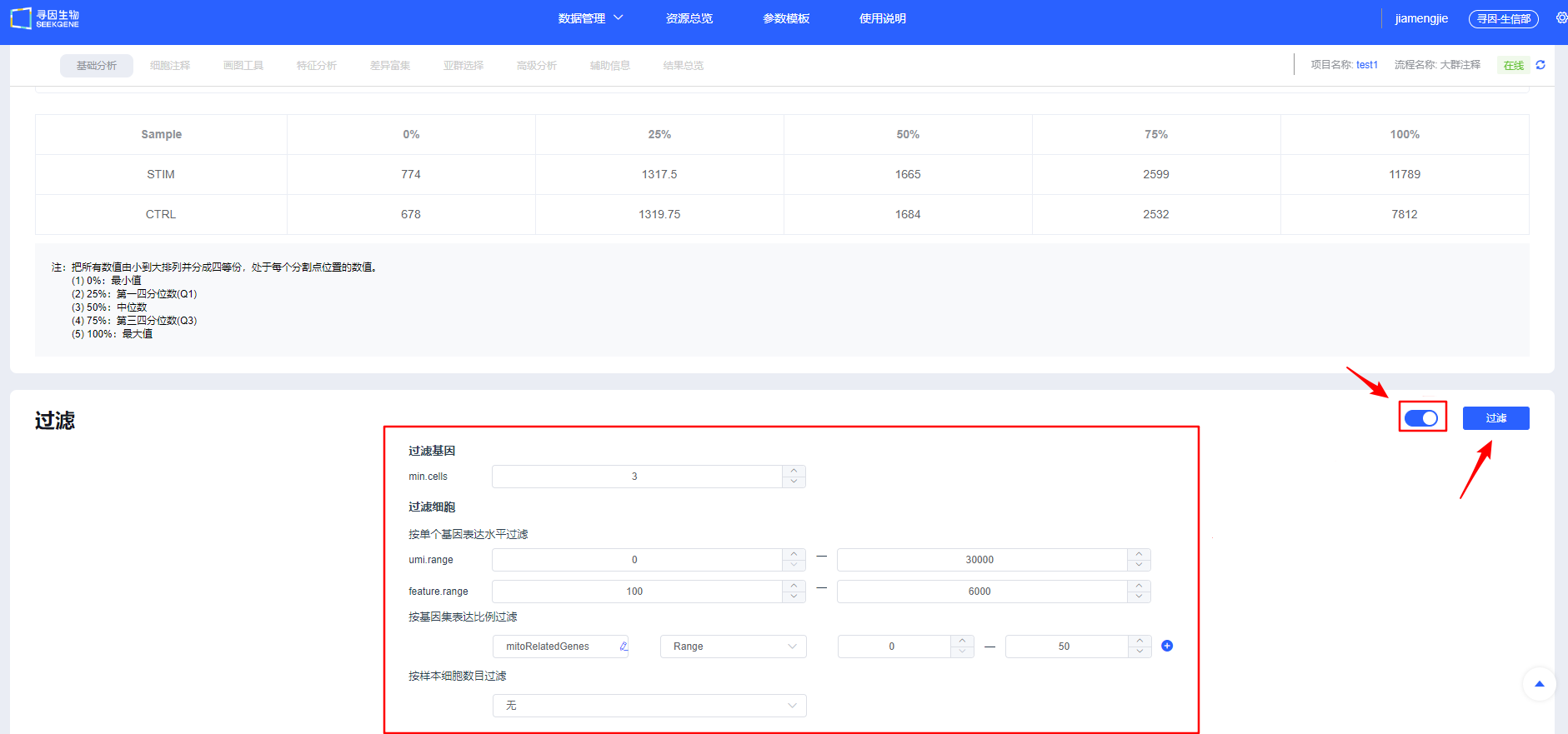
- 展开按钮可查看默认参数,用户可参考已发表单细胞文章方法中的过滤参数进行【过滤】。
TIP
如果选择的是已经整合的数据,点击【过滤】会提示是否需要跳过过滤整合步骤。如果不需要调整参数可跳过进行后续分析,如需调整过滤阈值则取消跳过,重新进行过滤整合。
IMPORTANT
过滤策略建议:
- 使用 MAD 动态阈值过滤 mt% 或以 10%-20% 作为参考上限;
- nCount_RNA/nFeature_RNA 以分位数或箱线图上界识别极端异常;
- 多样本时可按样本细胞数做均衡抽样,避免样本量主导聚类;
- 高代谢组织(心/肾/肝等)与免疫/粒细胞丰富样本需适当放宽阈值,避免误删真实细胞。
【过滤】后可查看各样本过滤后质量信息,同时可对单样本进行个性化调整,保证整体样本质量一致。
CAUTION
避免机械性依赖“拉高细胞数”的操作(如仅凭经验值强行放宽下限)。若瀑布图拐点不清、背景高或低 UMI 群占比异常,强行回收会降低下游稳定性。 双胞(doublet)排查思路:
- 识别 nCount_RNA/nFeature_RNA 上尾细胞(分布右侧极端高值);
- 是否存在互斥 marker 共表达、UMAP 两团之间的“桥状”细胞;
- 肿瘤项目可结合 CNV 辅助判定;
- 谨慎过滤:建议多证据一致时再剔除,并在剔除后重做整合与聚类验证一致性。
整合
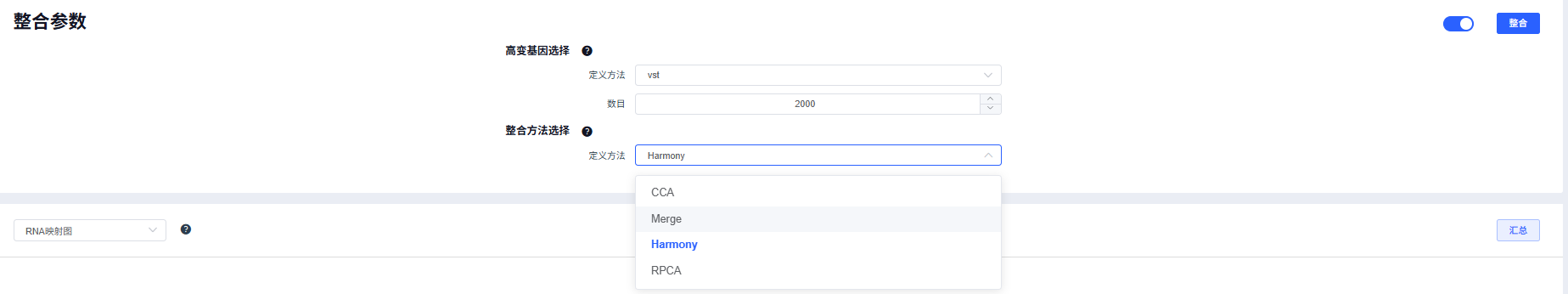
质控合格的数据进行【整合】,目前提供四种整合方法,其中CCA、Harmony和RPCA会对多个样本进行批次矫正。
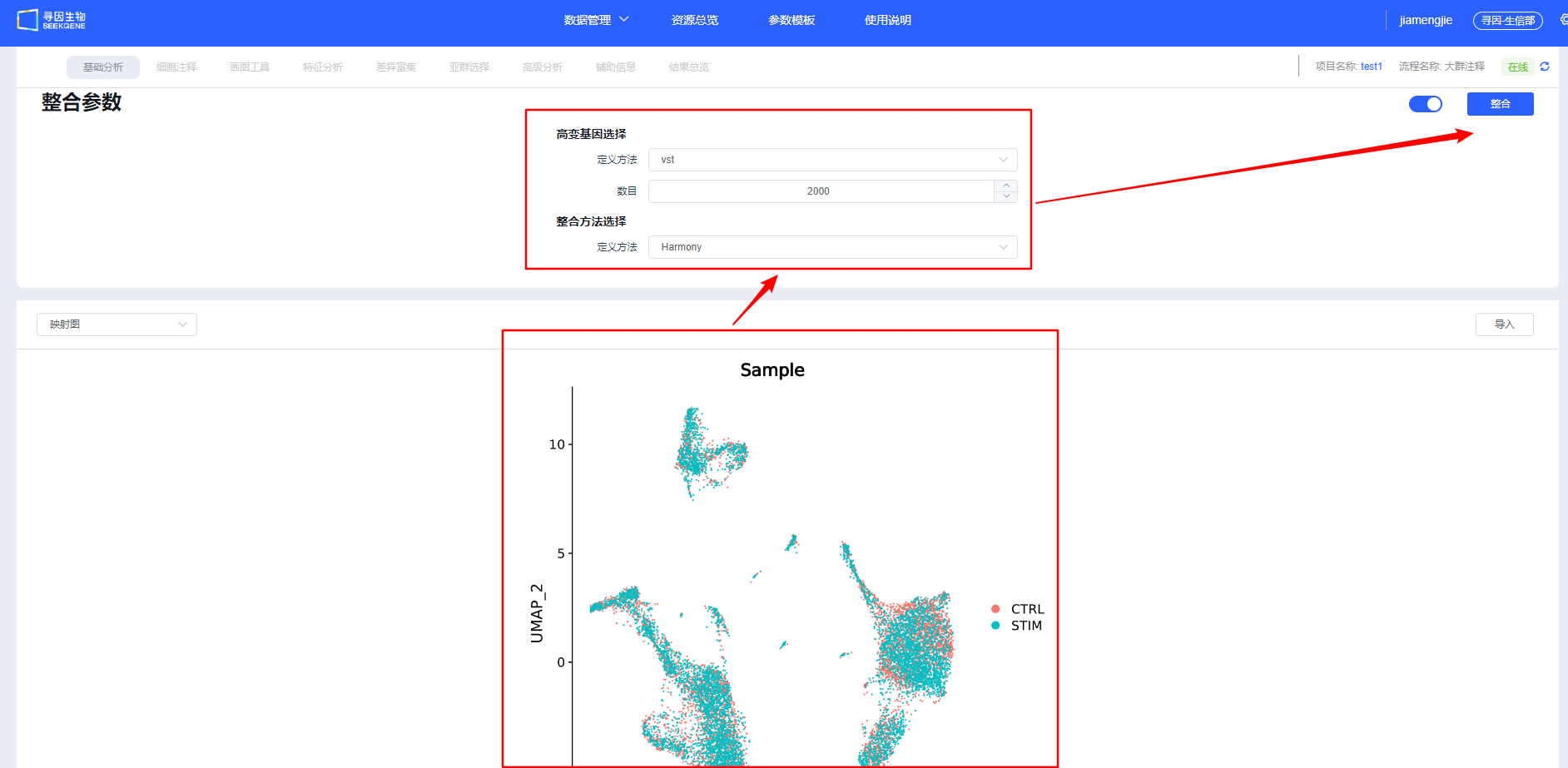
【整合】后会展示样本整合情况,可调整整合参数重新整合。建议用户尝试多种整合方法,选择更合适的方法进行后续分析。
TIP
整合方法选择:
- 批次效应较弱:merge 直接合并,避免过度校正;
- 中等批次:CCA/RPCA;
- 批次显著或异构明显:Harmony 更稳健。
TIP
效果评估三准则:
- 批次混合度:同一细胞类型在 UMAP/TSNE 中跨样本均匀混合;
- 生物信号保留:经典 marker 梯度与分群边界清晰,差异/富集结果符合预期;
- 过度/欠校正告警:过度校正会抹平差异,欠校正会出现“按批次聚类”。
聚类
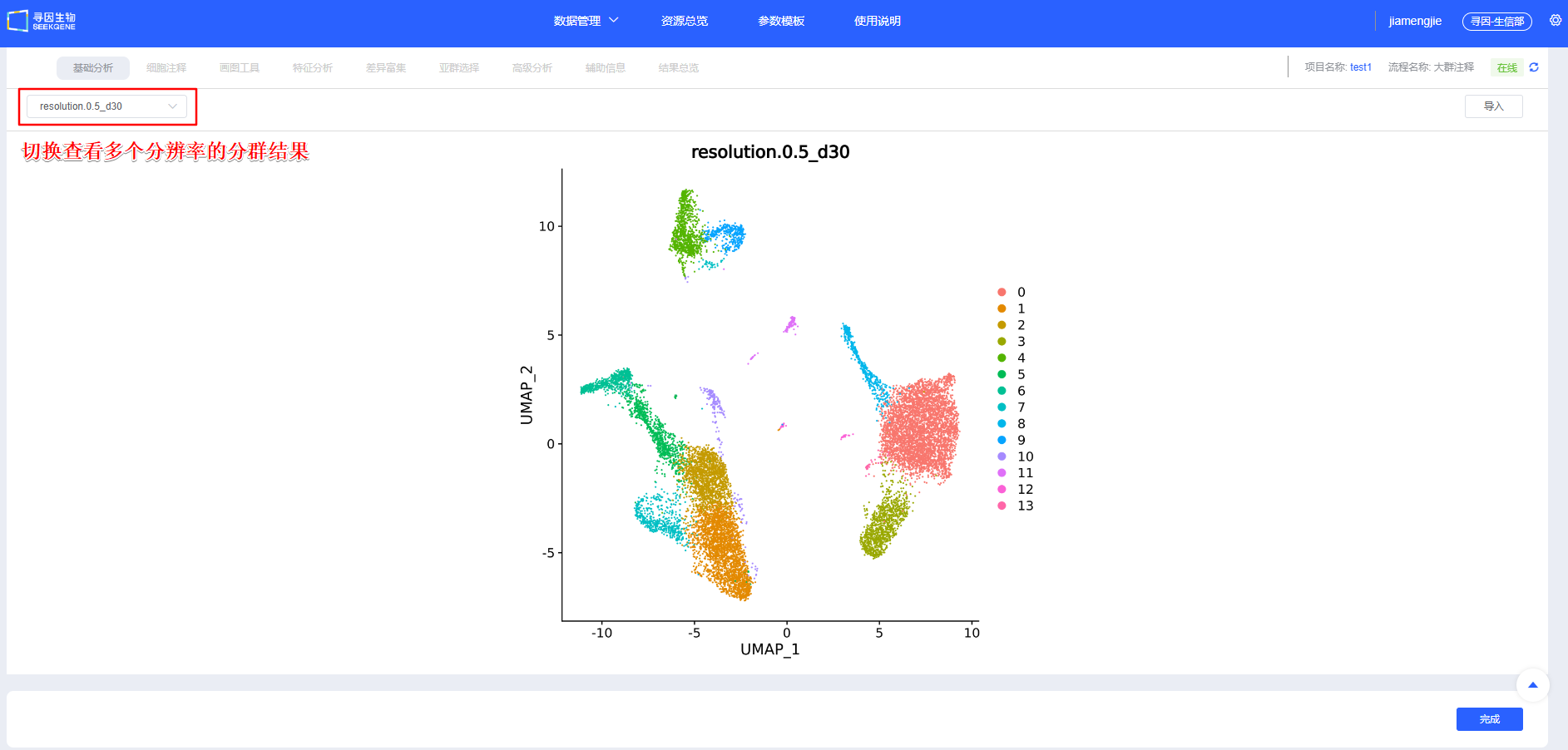
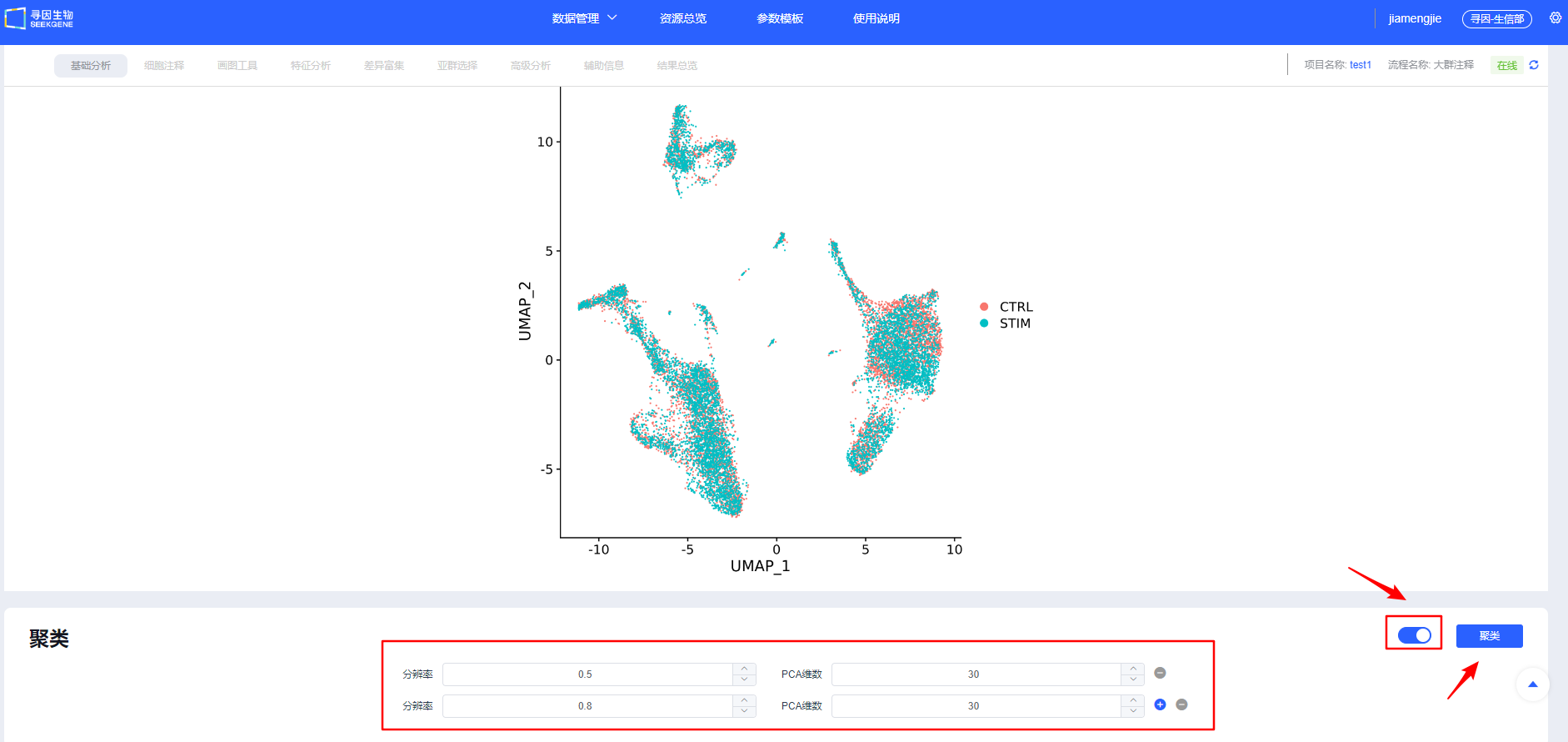
【整合】确认后进行【聚类】,可新建选择多个分辨率进行聚类,分辨率越大分群数量越多。后续模块也可新增分辨率进行聚类。
TIP
聚类调参与排错:
- 拐点法:以 PCA 肘部拐点作为 dims 起点,细胞量越大适当增加;
- 子集重聚类:如 T 细胞等大类单独重聚类,放大类内异质性。 dims 过低会遗漏关键异质性,过高易过聚类与放大噪音。请结合拐点与重现性优选。
分析完成
【聚类】后无需调整则点击【完成】,跳转“细胞注释”模块,正式开始进行单细胞相关分析。该步会耗费一定时间,请用户耐心等待。