CytoTRACE拟时序分析文档
前言
TIP
CytoTRACE (Cellular Trajectory Reconstruction Analysis using gene Counts and Expression) 是一种强大的、无需先验知识的拟时序分析工具。它通过评估单个细胞的转录多样性,能够稳健地预测细胞的分化状态,尤其擅长识别发育轨迹的起点(即分化潜能最高的细胞)。
在复杂的生物学系统中,如何确定哪个细胞群是最原始的“根”状态,是拟时序分析中的核心挑战。CytoTRACE基于一个核心假设:分化潜能越高的细胞(如干细胞、祖细胞),其转录活动越复杂,即可检测到的表达基因数量越多。 这一特性使其成为Monocle等轨迹构建工具的理想搭档,能够为轨迹的起点选择提供客观、无偏的证据。
CytoTRACE的核心功能
- 无偏的起点识别:无需指定任何起点或终点,自动预测细胞的分化潜能。
- 稳健性与普适性:在多种组织、物种和测序平台上得到广泛验证。
- 易于解读:提供一个从0(分化终点)到1(分化起点)的连续分数,直观反映细胞的分化程度。
本篇文档旨在为研究者提供一份详尽的CytoTRACE技术指南,内容涵盖其核心原理、在SeekSoulOnline云平台上的操作方法、结果解读、实战案例及常见问题,帮助您将其有效地整合到您的单细胞分析流程中。
CytoTRACE理论基础
核心原理
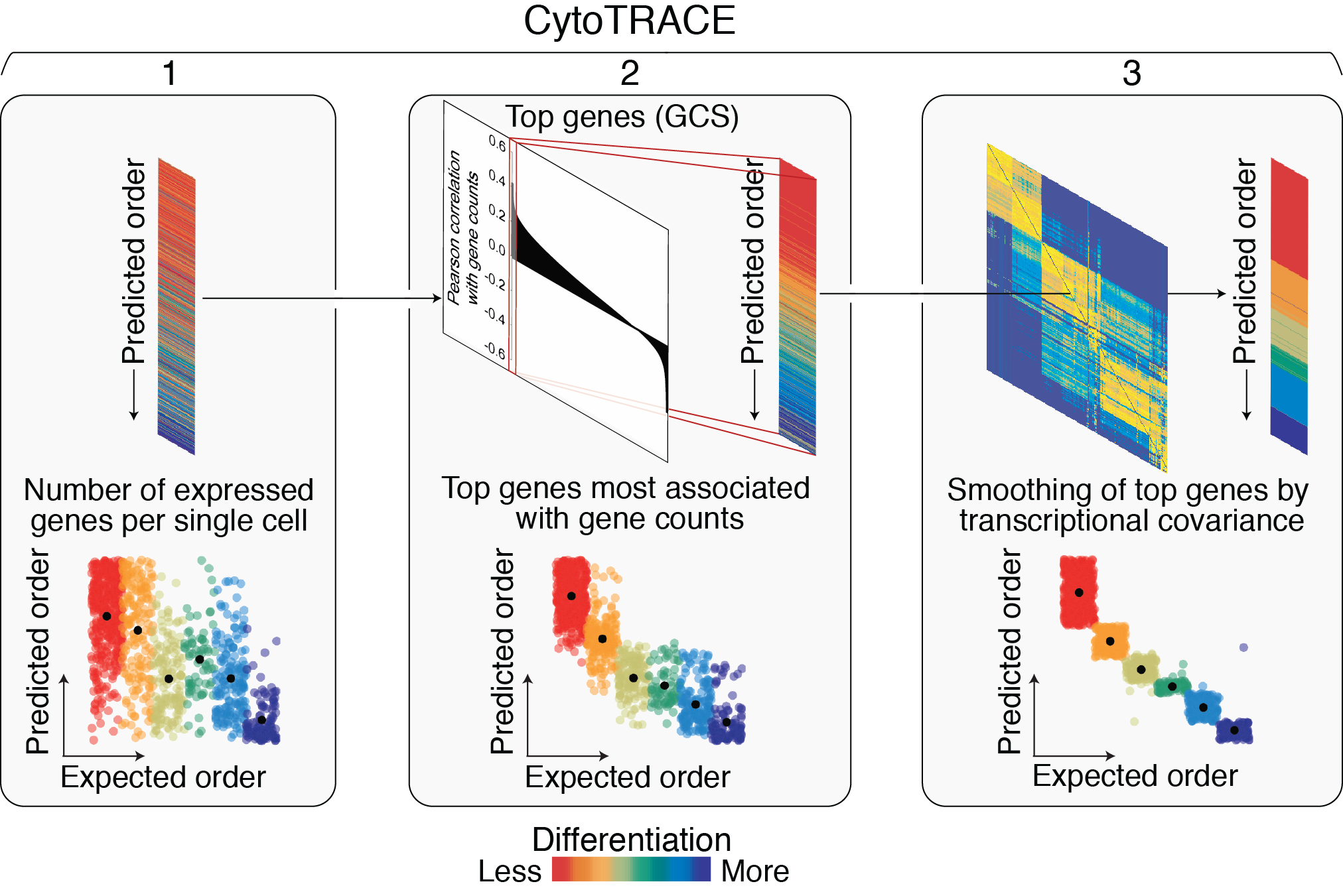
CytoTRACE的计算过程主要分为三个步骤:
基因计数 (Gene Counts):计算每个单细胞中表达量大于零的基因总数。这是衡量细胞转录复杂性的一个直接指标。
基因计数特征 (Gene Counts Signature, GCS):识别那些表达模式与基因计数正相关的基因。首先将表达矩阵标准化,然后计算每个基因与基因计数的相关性,最后选取相关性最强的前200个基因计算其几何平均表达值,得到GCS。
CytoTRACE分数计算:利用细胞间的局部相似性,通过两步平滑处理(马尔可夫模型及非负最小二乘回归)来迭代优化GCS,最终得到每个细胞的CytoTRACE分数。分数经过排序和缩放,范围在0到1之间,其中1代表分化程度最低(干性最高),0代表分化程度最高。
云平台操作指南
在seeksoulonline云平台上进行CytoTRACE分析时,您需要设置以下参数:
参数详解
| 界面参数 | 说明 |
|---|---|
| 任务名称 | 本次分析的任务名称,用于区分不同的分析任务。 |
| 分组因子 | 选择用于定义细胞群体的元数据列,通常是细胞类型注释或聚类结果。 |
| 细胞类型 | 选择要纳入分析的具体细胞亚群。 |
| 筛选因子 | 选择用于筛选样本或分组的元数据列,例如样本来源。 |
| 筛选对象 | 选择要保留用于分析的样本或分组。 |
| Downsample | 是否对细胞进行随机抽样以加速计算,适用于细胞数量庞大的数据集。 |
| Downsample_num | 如果选择抽样,这里设置抽样的细胞数量。 |
| 备注 | 对本次分析的自定义备注信息。 |
结果解读
分析完成后,平台会生成一份详细的报告,包含以下几个核心部分:
5.1 CytoTRACE分数映射图
这是最直观的结果,将CytoTRACE分数映射到UMAP/t-SNE等降维图上。
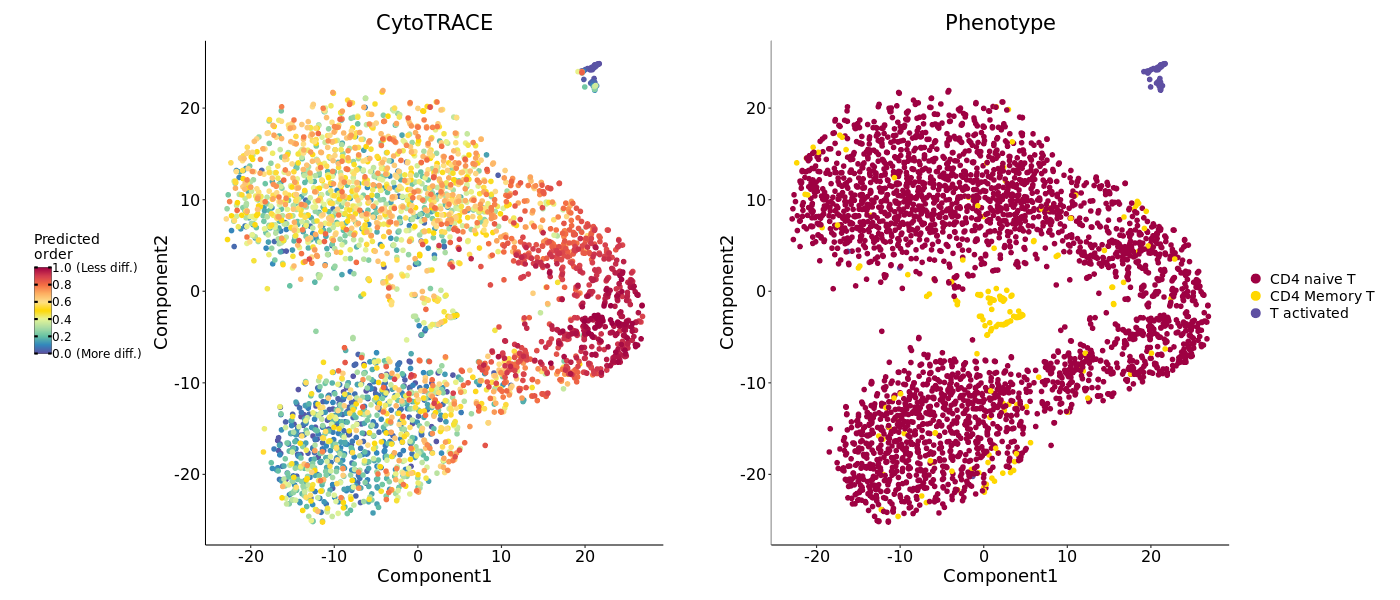
- 图表解读:每个点是一个细胞,颜色代表CytoTRACE分数。通常黄色代表高分(分化潜能高,接近起点),紫色代表低分(分化程度高,接近终点)。
- 分析要点:观察哪个细胞群颜色最亮(得分最高),这个细胞群就是轨迹的生物学起点。
5.2 分数箱线图
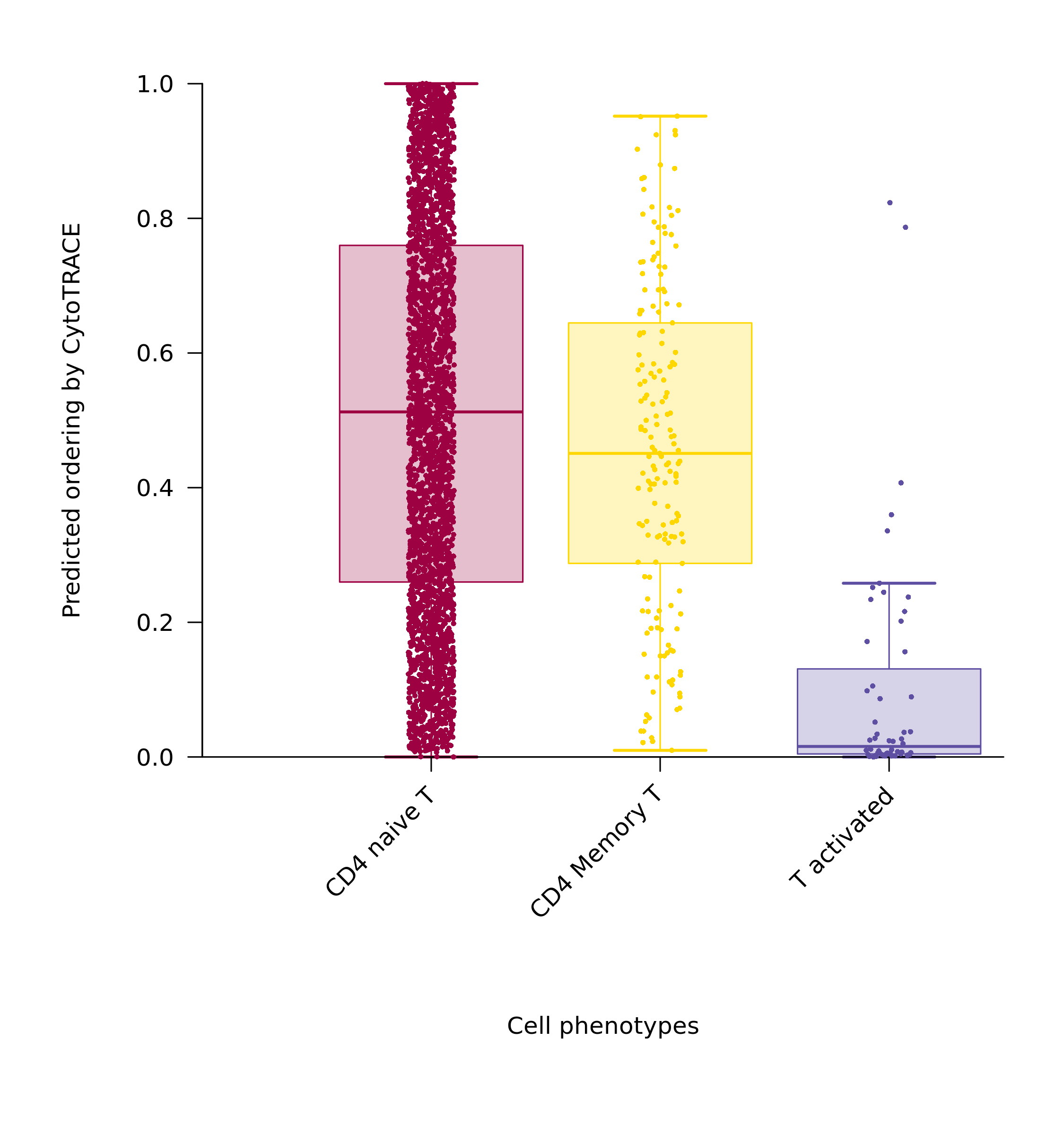
- 图表解读:按细胞类型(或您选择的分组)展示CytoTRACE分数的分布。
- 分析要点:定量比较不同细胞群体的分化潜能。例如,干细胞群的箱体位置应显著高于其他已分化细胞群。
5.3 CytoTRACE相关基因条形图
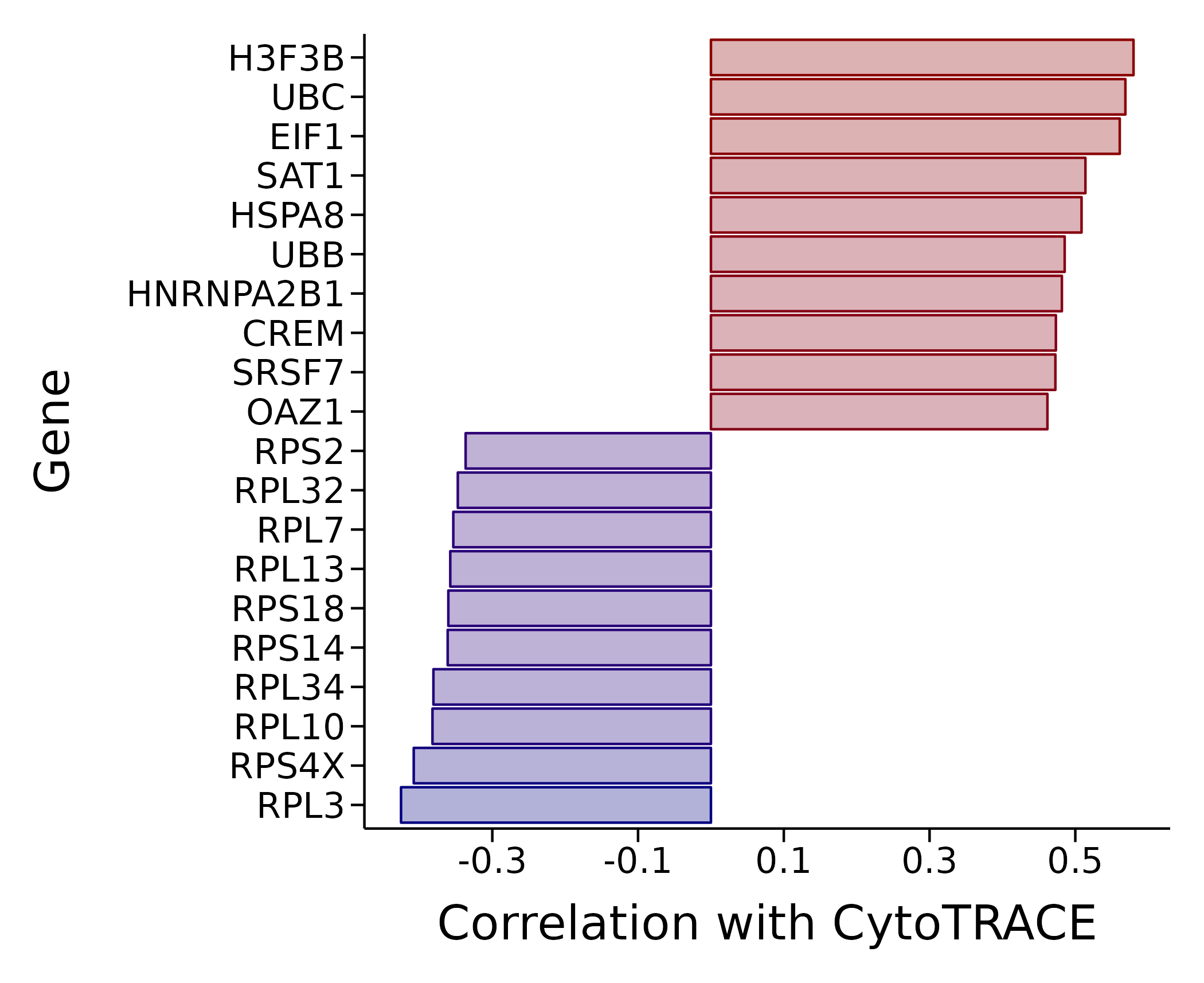
- 图表解读:该图通过条形图(Bar Plot)展示了与CytoTRACE分数相关性最强的基因。横轴代表相关性系数,纵轴是基因名称。
- 分析要点:
- 正相关基因(红色/暖色):条形朝右,表示这些基因的表达量与高分化潜能正相关。它们通常是与干性维持、祖细胞特性或细胞周期相关的基因。
- 负相关基因(蓝色/冷色):条形朝左,表示这些基因的表达量与低分化潜能(即高度分化)正相关。它们通常是细胞终末分化的功能性基因或标记基因。
- 通过分析这些基因,可以深入理解调控细胞分化状态的核心分子网络。
5.4 结果文件列表
核心数据文件:CytoTRACE_result.csv
这是分析最关键的输出文件,包含了每个细胞的详细评分信息:
| 列名 | 详细说明 |
|---|---|
| CytoTRACE | 预测的细胞分化潜能得分,范围从 1.0 (分化程度最低) 到 0.0 (分化程度最高)。 |
| CytoTRACErank | 根据 CytoTRACE 分数进行的排名。排名越高,代表细胞分化程度越低。 |
| GCS | 基因计数特征 (Gene Counts Signature),是与基因计数正相关的前200个基因的几何平均值。 |
| Counts | 每个细胞中检测到的表达基因总数 (Gene Counts)。 |
主要图表与文件
| 文件名 | 内容说明 |
|---|---|
CytoTRACE_result.csv | 上述核心数据文件。 |
CytoTRACE_plot_table.txt | 用于绘制分数箱线图的绘图数据。 |
CytoTRACE_plot.png/pdf | CytoTRACE分数在降维图上的映射图。 |
CytoTRACE_by_Phenotype_Boxplot.png/pdf | 按细胞类型展示的分数箱线图。 |
CytoGenes.png/pdf | 与CytoTRACE分数正负相关的Top基因条形图。 |
应用案例
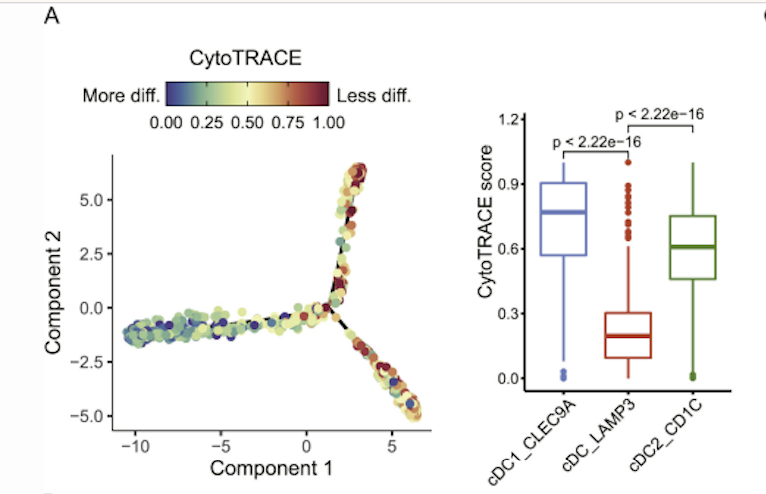
文献来源: A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021
研究背景: 该研究希望探究肿瘤浸润树突状细胞(DC)中一个成熟的LAMP3+ DC亚群的发育起源。
分析方法: 研究人员利用CytoTRACE评估了不同DC亚群的分化状态。结果显示,经典的cDC1和cDC2亚群相比于成熟的LAMP3+ DC亚群具有更高的CytoTRACE分数,表明它们处于更早的分化阶段。这一结果为LAMP3+ cDCs可能由cDC1和cDC2分化而来的假设提供了有力证据。
注意事项
1. CytoTRACE不是轨迹构建工具:它本身不生成细胞的连接路径或分支,而是为每个细胞提供一个“分化潜能”的度量。它的最佳用途是与其他工具(如Monocle2/3)结合,为其提供客观的起点信息。
2. 警惕技术性偏差:CytoTRACE的核心是基因计数,因此结果对测序深度和批次效应敏感。如果不同细胞类型在不同批次测序,或测序深度差异巨大,可能会引入技术偏差。在解释结果时需考虑这些因素。
3. 先分群,再分析:如果您的数据集包含生物学上完全不相关的细胞谱系(如免疫细胞和上皮细胞),建议先将它们分离,然后对您感兴趣的谱系(如T细胞分化谱系)单独运行CytoTRACE,结果会更有意义。
常见问题解答(FAQ)
Q1: 为什么应该使用 CytoTRACE?
A: CytoTRACE 可以在没有任何先验知识的情况下预测细胞分化状态,特别适用于以下场景:
- 在发育层次尚不明确的人类组织中预测分化状态。
- 在癌症等病变组织中研究细胞分化异质性。
- 为Monocle等轨迹构建工具提供客观、无偏的起点建议。
- 鉴定与干细胞特性和分化过程相关的关键基因。
Q2: 在哪些情况下 CytoTRACE 可能不适用或需要额外注意?
A: 在以下几种情况,建议进行额外的预处理或结果解读时需特别注意:
- 包含多种异质组织的数据集: CytoTRACE 会对数据集中所有细胞进行排序。如果数据来自完全不同的组织系统(如造血细胞和内皮细胞混合),建议先将其拆分再分别运行CytoTRACE。
- 包含静息和增殖干细胞群的数据: CytoTRACE本身可能难以区分这两种状态。可以结合总RNA含量等指标进行区分,通常静息干细胞的RNA含量较低。
- 强烈的批次效应: 如果不同细胞类型在不同批次中测序,可能会引入技术差异导致的基因计数偏差,从而影响结果。实验设计时应尽量避免此类批次效应。
参考文献
- Gulati, G. S., et al. (2020). Single-cell transcriptional diversity is a hallmark of developmental potential. Science, 367(6476), 405–411.
- Cheng S, et al. (2021). A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell, 184(3):792-809.e23.