“基础分析”是分析流程的初始模块,可进行基因、线粒体过滤、去批次整合聚类,保留质量合格的细胞用于后续分析。分析流程创建及过滤整合聚类流程如下:
初始阶段
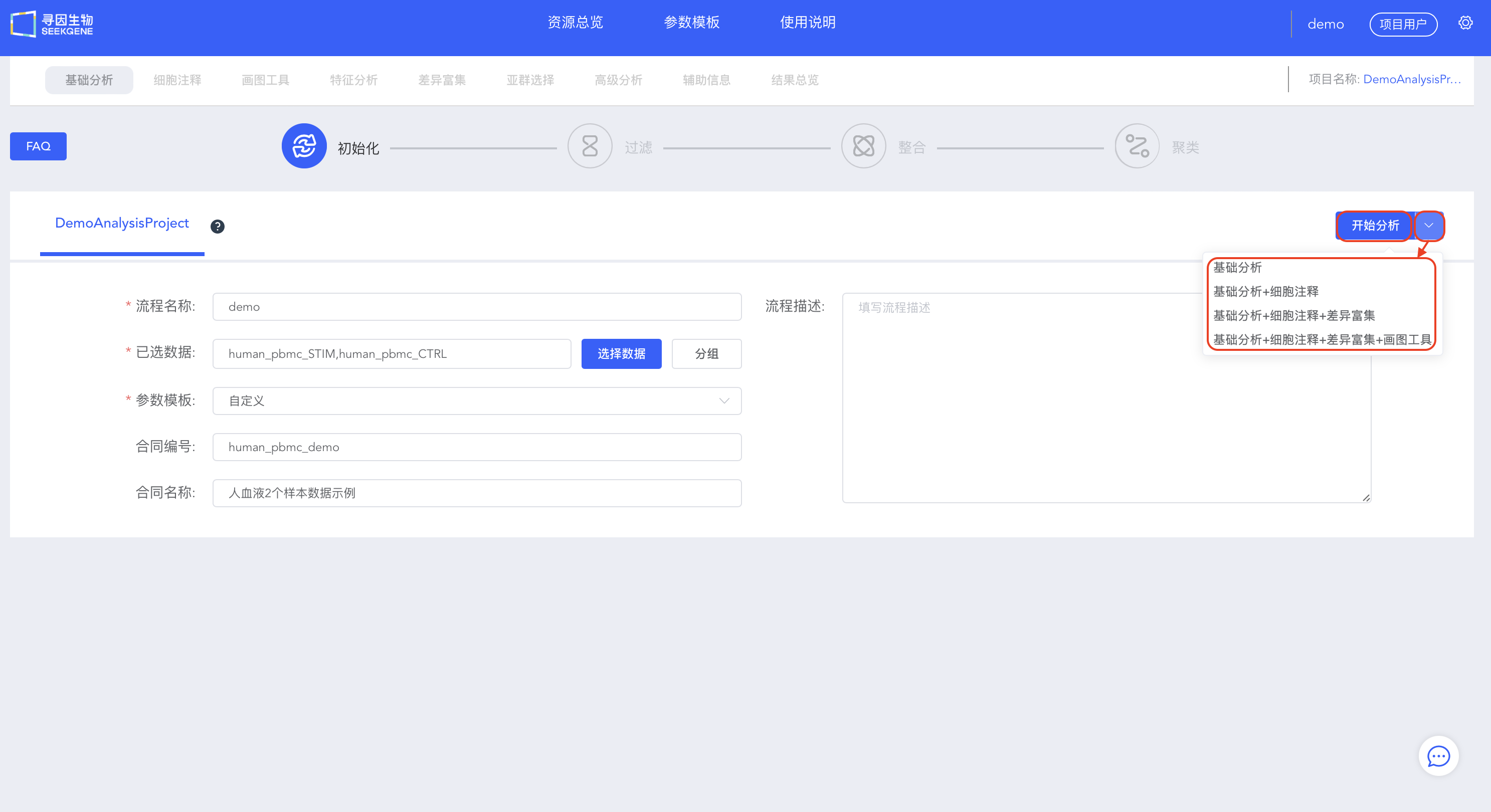
【新建流程】创建单细胞分析流程。项目开始一般选择样本进行大群分析,后续可选择注释好的细胞类型进行亚群分析。
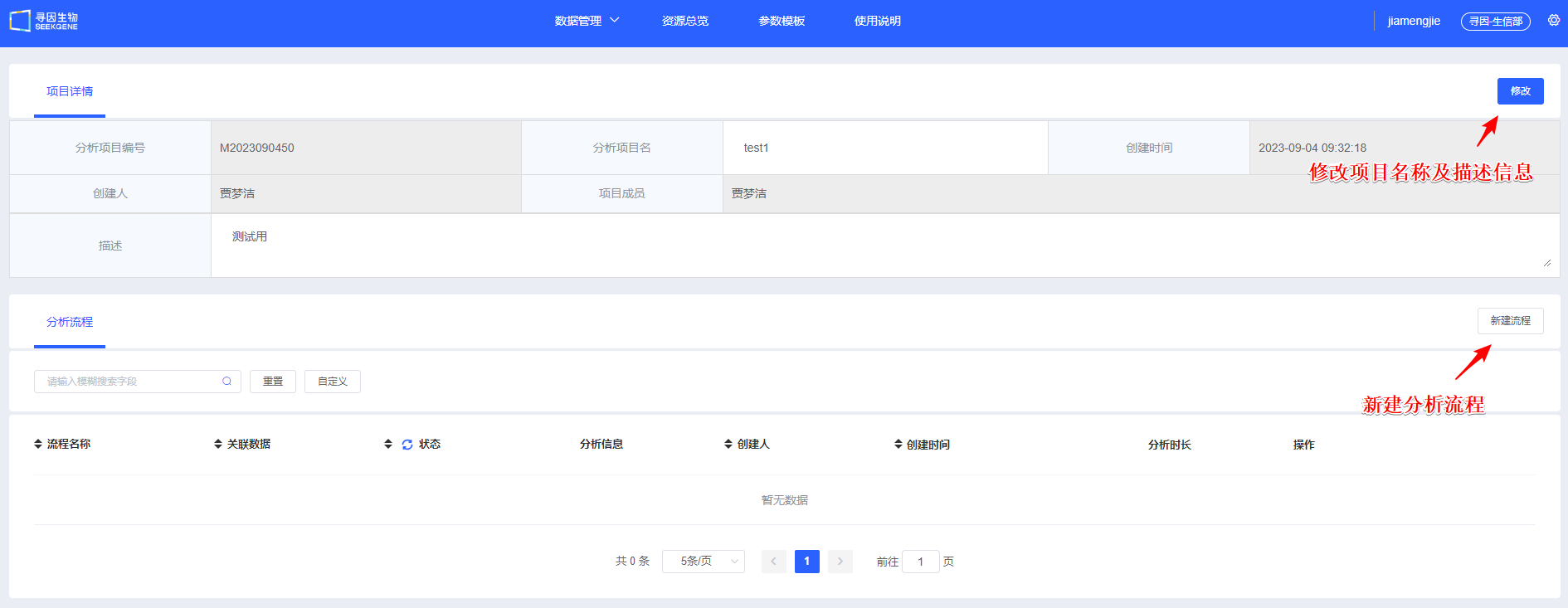
填写“流程名称”及“流程描述”,用于后续查找和了解流程信息。
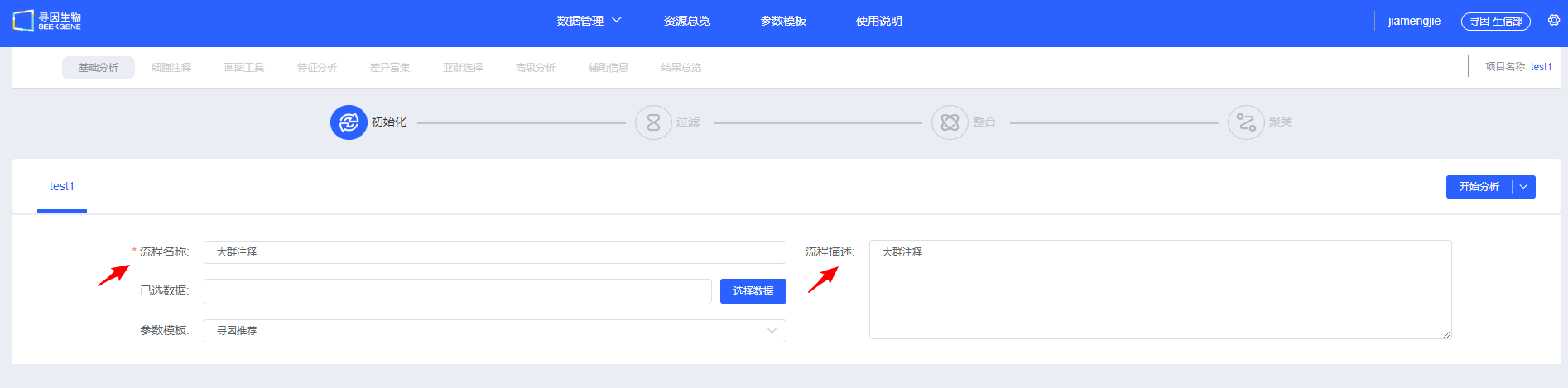
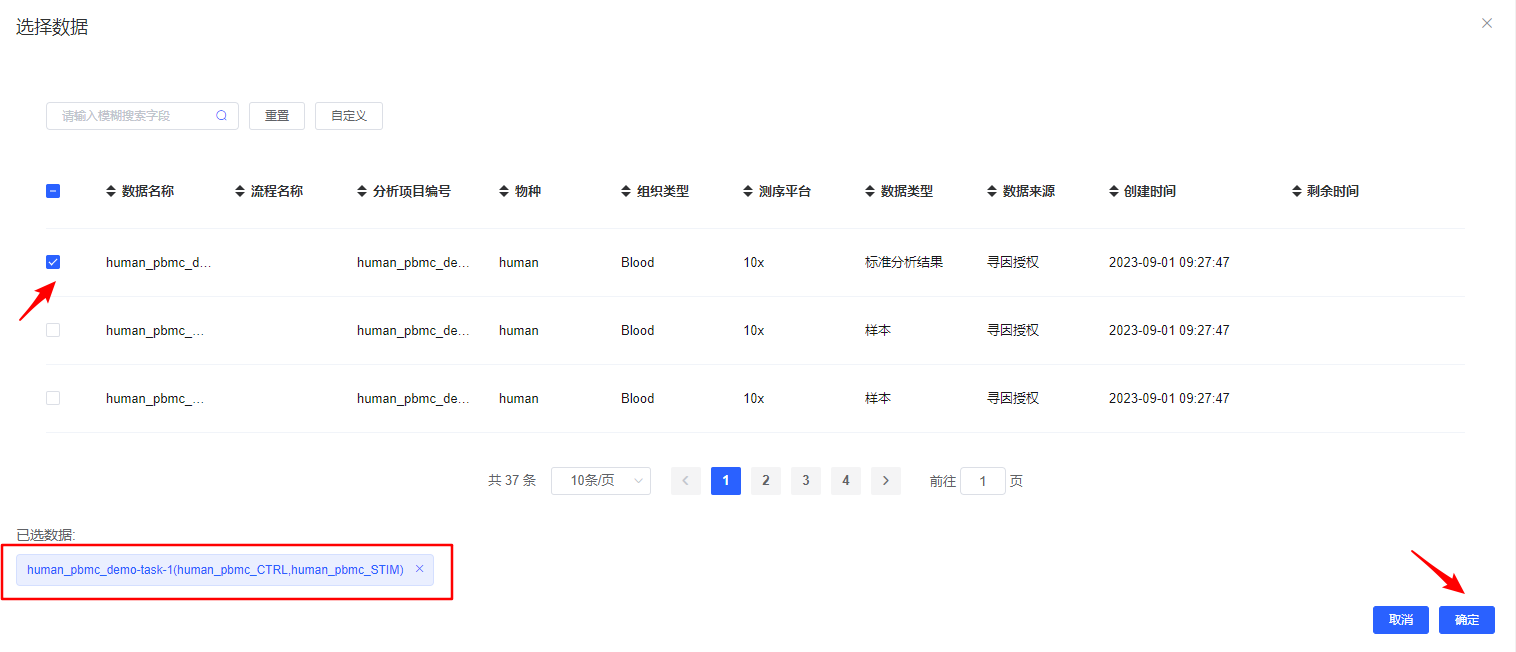
【选择数据】选择待分析的样本,可选择已经整合好的多样本数据,也可选多个单样本进行过滤整合。
NOTE
数据详细来源可查看《我的数据》。
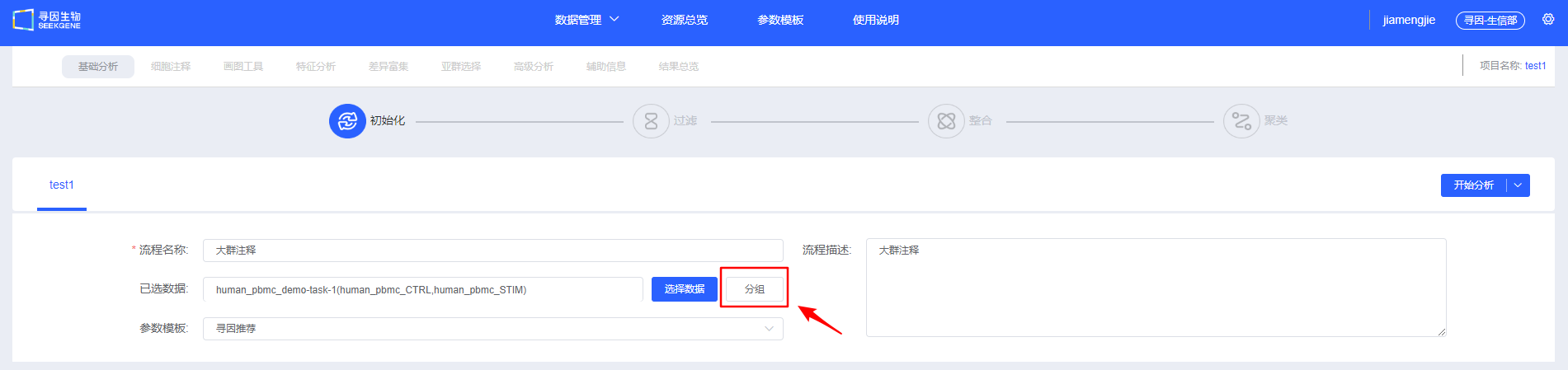
- 填写信息,选择待分析的数据,可以选择【开始分析】手动进行过滤整合和聚类,也可以选择【基础分析】、【基础分析+细胞注释】、【基础分析+细胞注释+差异富集】或【基础分析+细胞注释+差异富集+画图工具】快速进行自动分析。
过滤
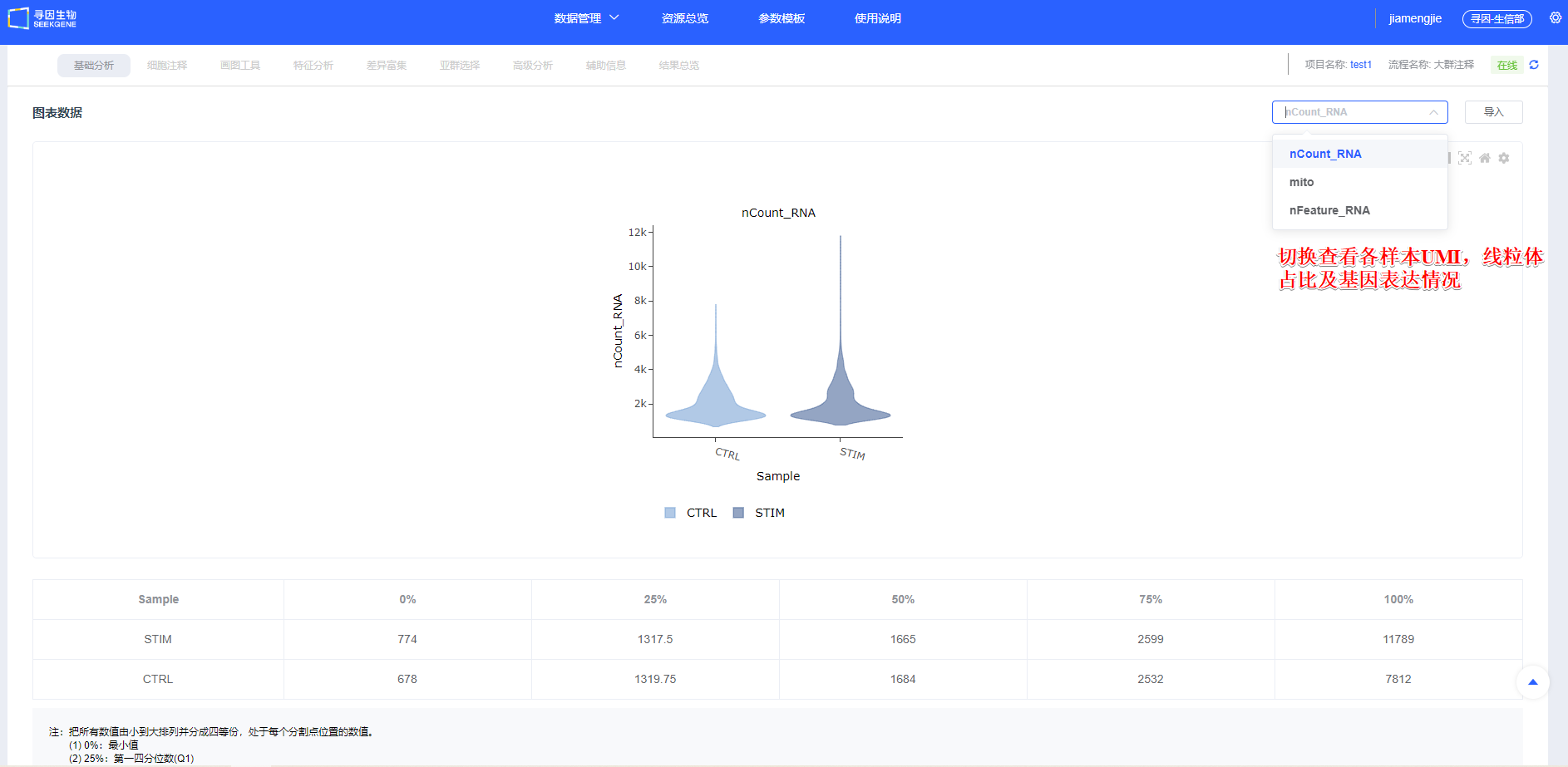
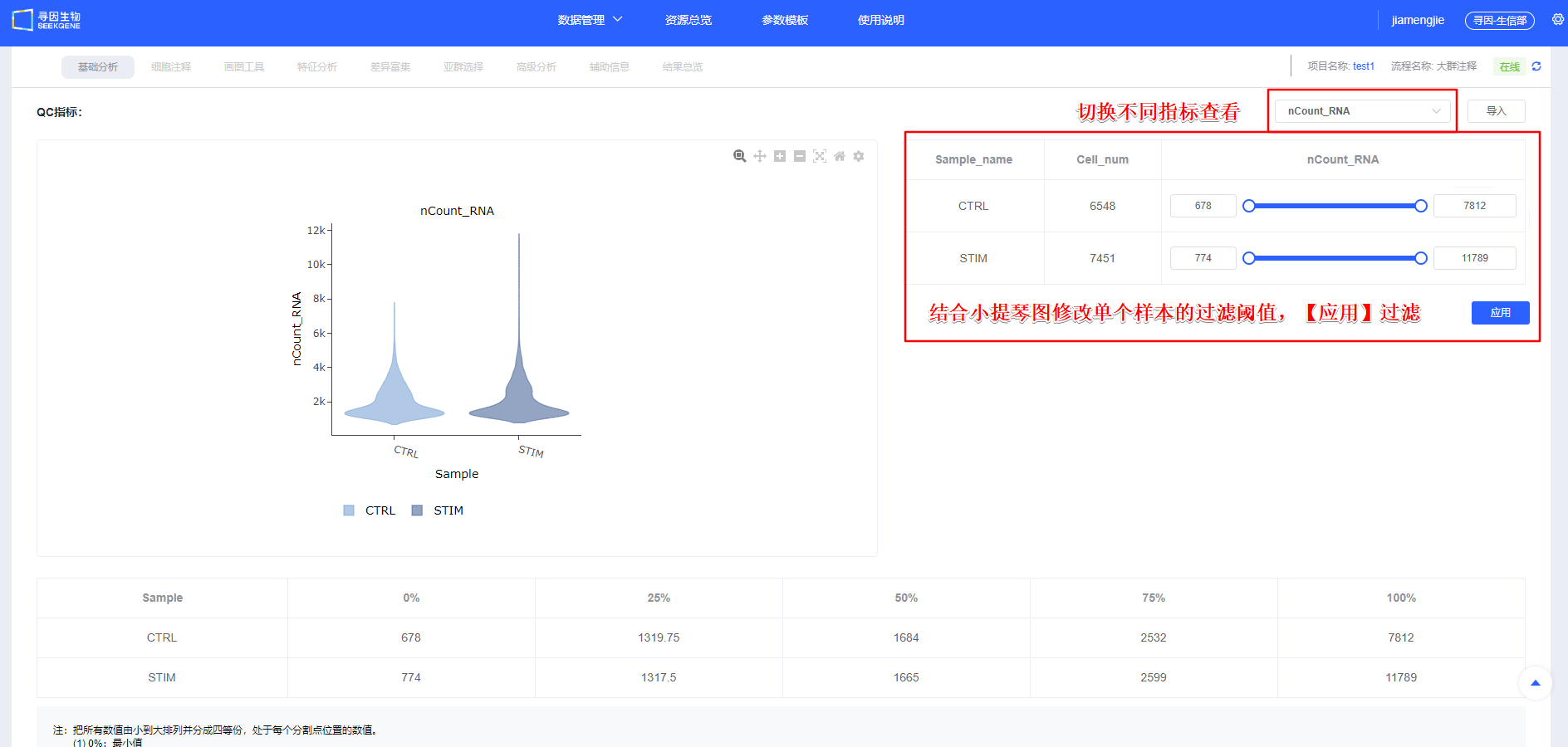
- 【开始分析】后的“图表数据”会统计样本UMI、线粒体占比及基因表达情况,简要展示各样本质量信息。
NOTE
指标释义(scRNA-seq):
- nCount_RNA:每细胞 UMI 总数,反映测序深度与转录本丰度
- nFeature_RNA:每细胞检出的基因数,反映表达复杂度
- 线粒体比例(mito):线粒体基因 UMI 占比,偏高常与凋亡/损伤相关(高代谢组织可适度放宽)
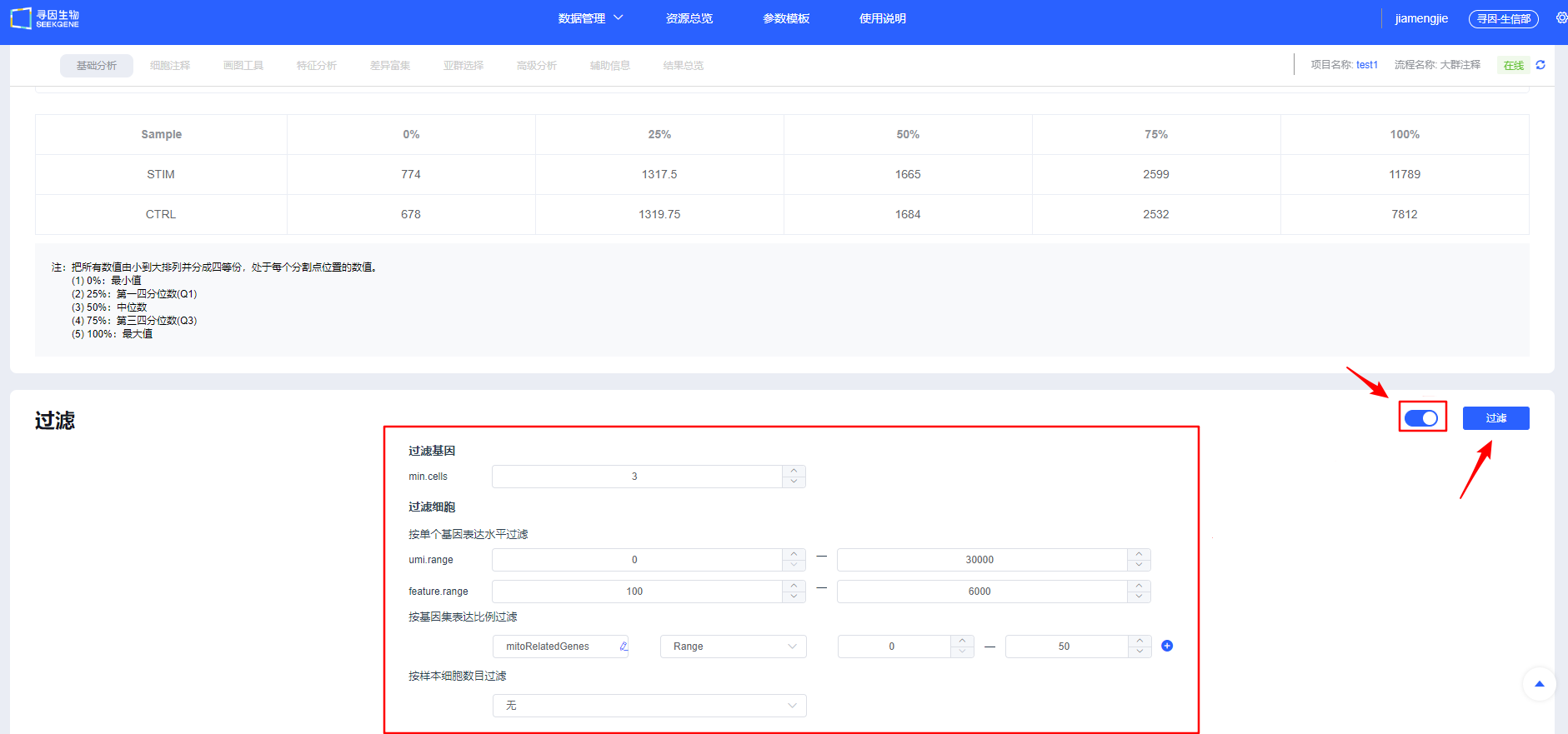
- 展开按钮可查看默认参数,用户可参考已发表单细胞文章方法中的过滤参数进行【过滤】。
TIP
如果选择的是已经整合的数据,点击【过滤】会提示是否需要跳过过滤整合步骤。如果不需要调整参数可跳过进行后续分析,如需调整过滤阈值则取消跳过,重新进行过滤整合。
IMPORTANT
过滤策略建议:
- 使用 MAD 动态阈值过滤 mt% 或以 10%-20% 作为参考上限;
- nCount_RNA/nFeature_RNA 以分位数或箱线图上界识别极端异常;
- 多样本时可按样本细胞数做均衡抽样,避免样本量主导聚类;
- 高代谢组织(心/肾/肝等)与免疫/粒细胞丰富样本需适当放宽阈值,避免误删真实细胞。
【过滤】后可查看各样本过滤后质量信息,同时可对单样本进行个性化调整,保证整体样本质量一致。
CAUTION
避免机械性依赖“拉高细胞数”的操作(如仅凭经验值强行放宽下限)。若瀑布图拐点不清、背景高或低 UMI 群占比异常,强行回收会降低下游稳定性。 双胞(doublet)排查思路:
- 识别 nCount_RNA/nFeature_RNA 上尾细胞(分布右侧极端高值);
- 是否存在互斥 marker 共表达、UMAP 两团之间的“桥状”细胞;
- 肿瘤项目可结合 CNV 辅助判定;
- 谨慎过滤:建议多证据一致时再剔除,并在剔除后重做整合与聚类验证一致性。
整合
质控合格的数据进行【整合】,目前提供四种整合方法,其中CCA、Harmony和RPCA会对多个样本进行批次矫正。
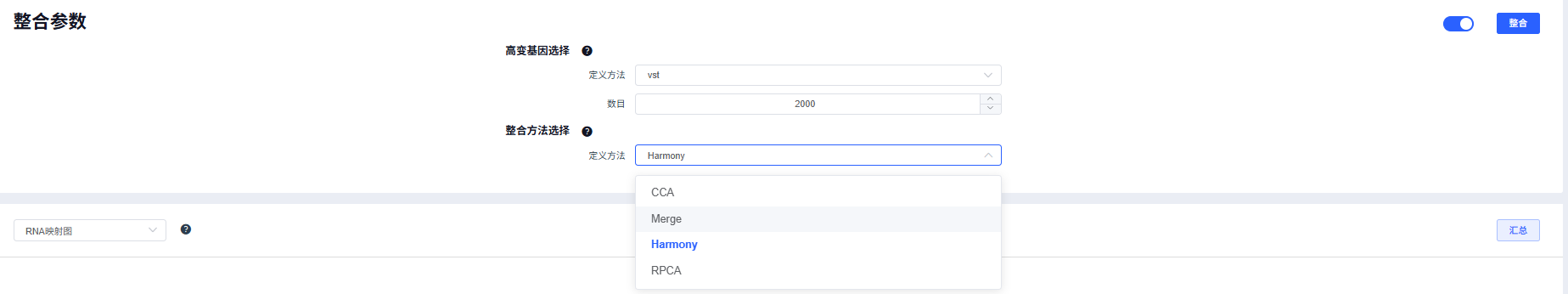
【整合】后会展示样本整合情况,可调整整合参数重新整合。建议用户尝试多种整合方法,选择更合适的方法进行后续分析。
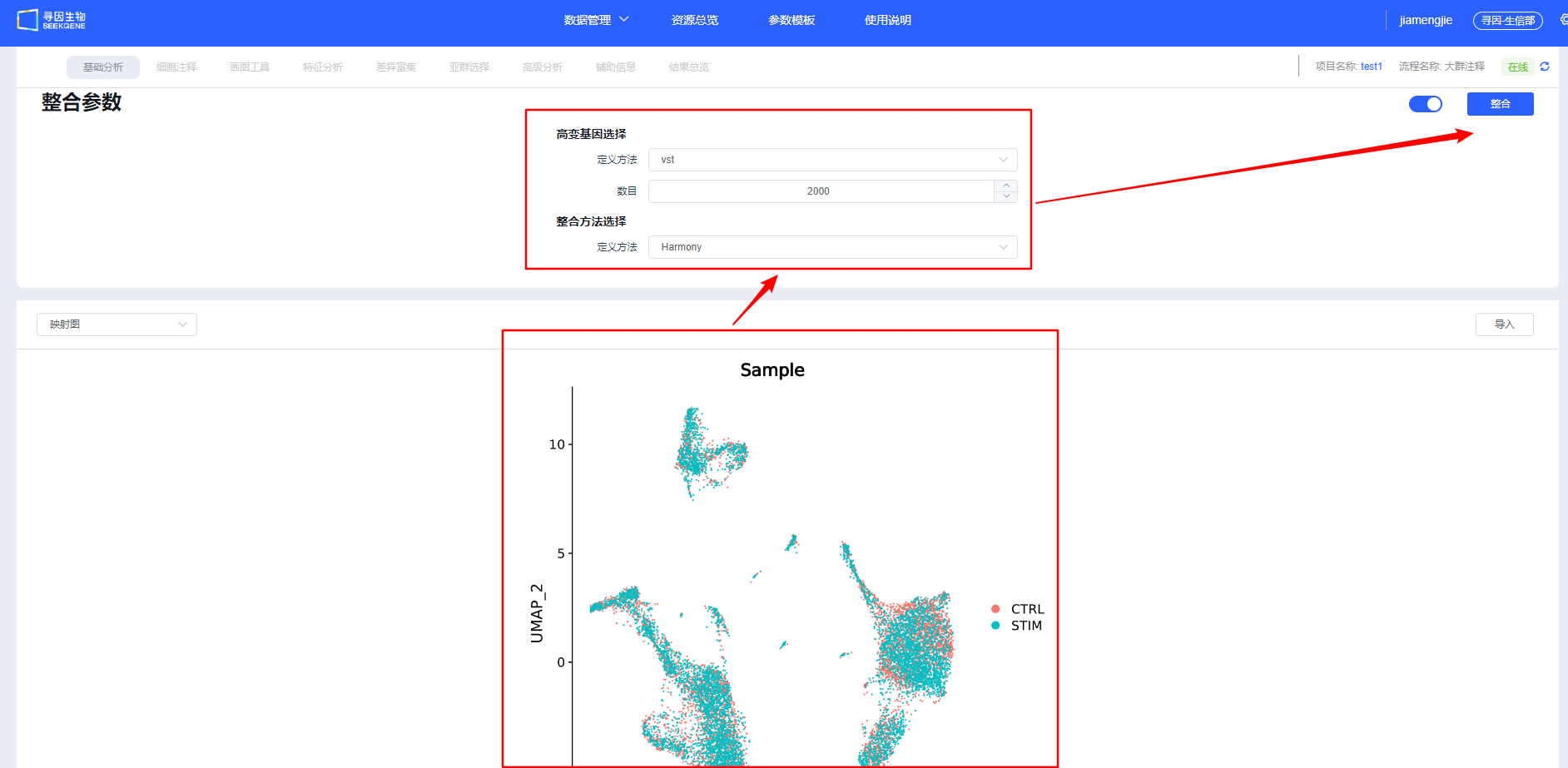
TIP
整合方法选择:
- 批次效应较弱:merge 直接合并,避免过度校正;
- 中等批次:CCA/RPCA;
- 批次显著或异构明显:Harmony 更稳健。
TIP
效果评估三准则:
- 批次混合度:同一细胞类型在 UMAP/TSNE 中跨样本均匀混合;
- 生物信号保留:经典 marker 梯度与分群边界清晰,差异/富集结果符合预期;
- 过度/欠校正告警:过度校正会抹平差异,欠校正会出现“按批次聚类”。
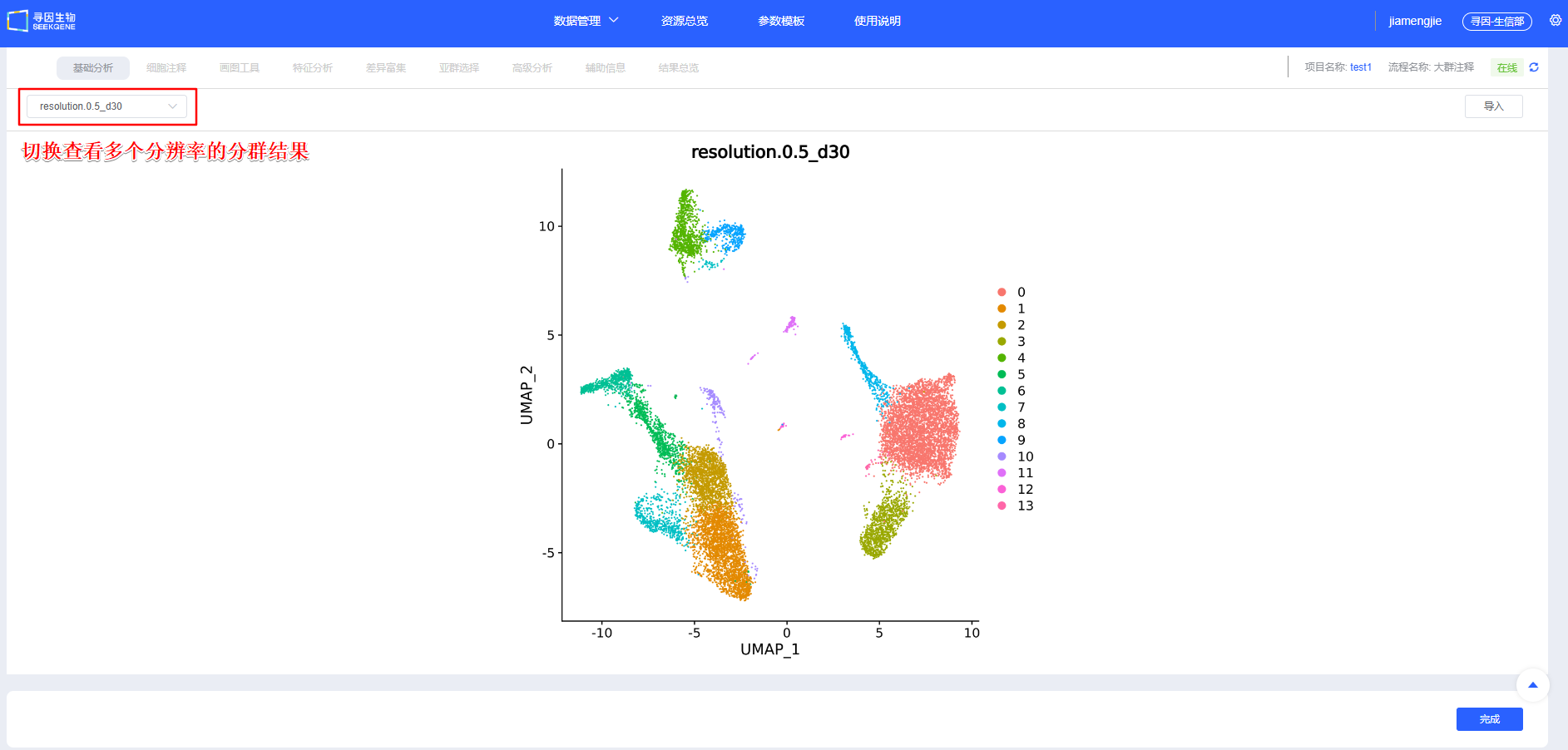
聚类
【整合】确认后进行【聚类】,可新建选择多个分辨率进行聚类,分辨率越大分群数量越多。后续模块也可新增分辨率进行聚类。
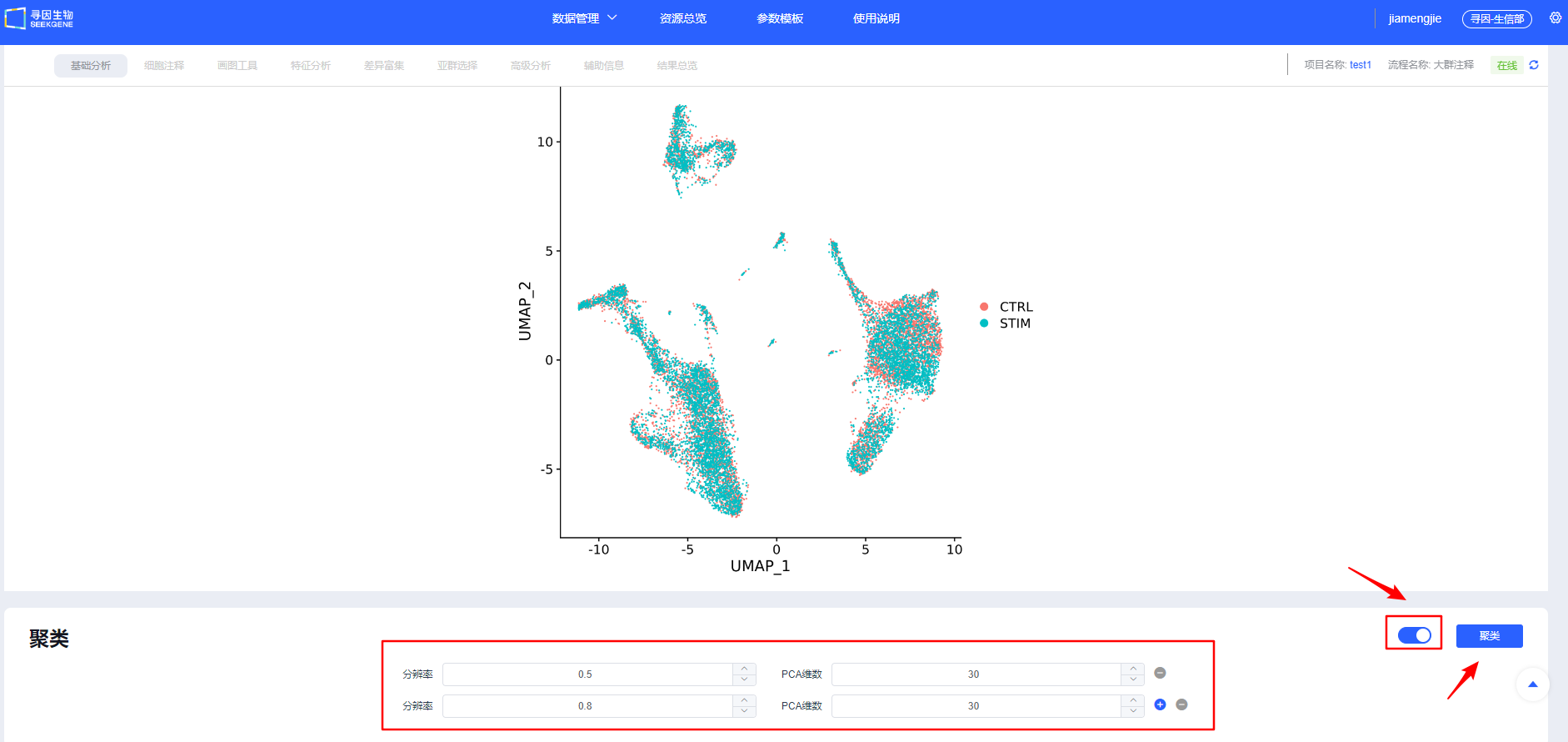
TIP
聚类调参与排错:
- 拐点法:以 PCA 肘部拐点作为 dims 起点,细胞量越大适当增加;
- 子集重聚类:如 T 细胞等大类单独重聚类,放大类内异质性。 dims 过低会遗漏关键异质性,过高易过聚类与放大噪音。请结合拐点与重现性优选。
分析完成
【聚类】后无需调整则点击【完成】,跳转“细胞注释”模块,正式开始进行单细胞相关分析。该步会耗费一定时间,请用户耐心等待。
“基础分析”是分析流程的初始模块,可进行基因、线粒体过滤、去批次整合聚类,保留质量合格的细胞用于后续分析。分析流程创建及过滤整合聚类流程如下:
初始阶段
【新建流程】创建单细胞分析流程。项目开始一般选择样本进行大群分析,后续可选择注释好的细胞类型进行亚群分析。
填写“流程名称”及“流程描述”,用于后续查找和了解流程信息。
【选择数据】选择待分析的样本,可选择已经整合好的多样本数据,也可选多个单样本进行过滤整合,无论是整合好的还是单样本的,都需要选择ATAC标准分析结果的数据类型。

TIP
数据详细来源可查看《我的数据》。
- 填写信息,选择待分析的数据后,选择流程类型为scRNA+scATAC,可以选择【开始分析】手动进行过滤整合和聚类,也可以点击下拉框选择【基础分析】、【基础分析+细胞注释】、【基础分析+细胞注释+差异富集】或【基础分析+细胞注释+差异富集+画图工具】快速进行自动分析。

过滤
- 【开始分析】后的“图表数据”会统计样本RNA表达数据的UMI、线粒体占比及基因表达情况,也会统计ATAC数据的核小体信号、TSS富集分数和细胞中peaks等指标,简要展示各样本各个质量指标信息。

- 展开按钮可查看默认参数,用户可参考已发表单细胞文章方法中的过滤参数进行【过滤】。
TIP
如果选择的是已经整合的数据,点击【过滤】会提示是否需要跳过过滤整合步骤。如果不需要调整参数可跳过进行后续分析,如需调整过滤阈值则取消跳过,重新进行过滤整合。

【过滤】后可查看各样本过滤后质量信息,同时可对单样本进行个性化调整,保证整体样本质量一致。

WARNING
避免机械性依赖“拉高细胞数”的操作(如仅凭经验值强行放宽下限)。若瀑布图拐点不清、背景高或低 UMI 群占比异常,强行回收会降低下游稳定性。
详细scRNA-seq+scATAC-seq双组学数据质控相关的知识讲解可查看《单细胞ATAC_RNA多组学的质控》。
整合
质控合格的数据进行【整合】,目前提供四种整合方法,其中CCA、Harmony和RPCA会对多个样本进行批次矫正。在整合的时候,样本间的scRNA-seq数据和scRNA-seq数据进行整合,scATAC-seq数据和scATAC-seq数据进行整合。选择RNA数据的整合方法后,ATAC数据的整合方法会自动根据RNA的整合方法进行自匹配。

TIP
ATAC的min.cutoff参数默认值是5,该参数建议直接选择默认值

【整合】后可分别查看RNA数据和ATAC数据的整合情况,可调整整合参数重新整合。建议用户尝试多种整合方法,选择更合适的方法进行后续分析。

TIP
整合方法选择:
- 批次效应较弱:merge 直接合并,避免过度校正;
- 中等批次:Harmony,通常同平台产出的数据间的批次用Harmony就可有效矫正,CCA/RPCA可能会过度矫正;
- 批次显著或异构明显:CCA/RPCA 更稳健。
TIP
效果评估三准则:
- 批次混合度:同一细胞类型在 UMAP/TSNE(
检查RNA整合效果)、ATACUAMP/ATACTSNE(检查ATAC整合效果)中跨样本均匀混合; - 生物信号保留:经典 marker 梯度与分群边界清晰,差异/富集结果符合预期;
- 过度/欠校正告警:过度校正会抹平差异,欠校正会出现“按批次聚类”。
聚类
【整合】确认后进行【聚类】,可新建选择多个分辨率进行聚类,scRNA-seq和scATAC-seq双组学数据会进行三种聚类:基于ATAC数据进行聚类、基于RNA数据进行聚类、联合RNA和ATAC数据进行wnn聚类。后续模块也可新增分辨率进行聚类。

TIP
聚类调参
- RNA数据的拐点法:以 PCA 肘部拐点作为 dims 起点,细胞量越大适当增加;
- ATAC数据的lsi的第一个dim通常是技术噪音,需要从第2个lsi开始选择 dims 过低会遗漏关键异质性,过高易过聚类与放大噪音。请结合拐点与重现性优选。
详细scRNA-seq+scATAC-seq双组学数据降维聚类相关的知识讲解可查看《单细胞ATAC_RNA多组学的降维聚类》。
分析完成
【聚类】后无需调整则点击【完成】,跳转“细胞注释”模块,正式开始进行单细胞相关分析。该步会耗费一定时间,请用户耐心等待。
“基础分析”是分析流程的初始模块,可进行基因、线粒体过滤、去批次整合聚类,保留质量合格的细胞用于后续分析。分析流程创建及过滤整合聚类流程如下:
查看更多内容(展开/收起)
空间基础分析前言
细胞质控
细胞层面过滤(表达矩阵)
- 推荐使用 UMI 总数、检测到的基因数、线粒体比例三类指标联合筛选;根据样本组织类型自适应阈值。
- 在 SeekSpace 流程中,常用
--forceCell与--min_umi进行初筛(例如默认提取前80,000个UMI并过滤UMI<200的条形码)。如组织复杂或背景高噪时,适当提高--min_umi。
空间特异的条形码清洗(关键)
- 过滤“无效的spatial barcode”(表达文库短片段误入、测序错误等导致在HDMI库无位置信息的条形码)。
- 处理“重复且位置不一致”的spatial barcode:无法唯一定位者剔除。
- 清除“异常高支持”的空间条形码簇:将芯片按网格(如30×30像素bin)统计,识别并移除疑似脱片/飞溅造成的异常热点(优先剔除该bin中UMI最高的cell barcode对应的spatial barcode)。
- 最终仅保留通过细胞判定的cell barcode及其对应的有效spatial barcode。
唯一中心性检查(多中心细胞过滤)
- 基于细胞在空间上的UMI分布,定义“中心bin”与“核心区域”,计算核心与次中心的UMI比值(≥2判定为唯一中心)。
- 剔除多中心细胞,降低条形码混溢或核碎片导致的定位歧义。
空间映射
三库协同(以SeekSpace为例)
- 表达文库:用于纠错细胞条形码、提取UMI、生成表达矩阵。
- 空间文库:Spatai 文库 R2带有空间位置 32bp barcode :在R2上提取spatial barcode,并生成细胞标签(cell barcode)与空间标签(spatial barcode)的对应关系。空间文库的UMI(spatial UMI)代表了每个细胞标签上每个空间标签的表达量。
- HDMI文库:包含32bp空间条形码及其绝对坐标;获得的reads 前面是32bp 的空间barcode,后面是对应的空间坐标信息。
校正与配准流程
- 条形码层面:
- 细胞条形码/UMI纠错(允许/禁止错配可配置,如
--skip_misB等)。 - 空间条形码白名单化与纠错,绑定至HDMI坐标。
- 细胞条形码/UMI纠错(允许/禁止错配可配置,如
- 图像层面:
- 组织图像(DAPI/HE)进行缩放与模糊预处理,分割组织区域。
- 若自动配准不理想,可人工对齐并保存参数(如
parameters.json),再以“realign”流程复用,生成一致的背景掩膜与对齐图。
- 条形码层面:
空间坐标确立与细胞位置判定
- 依据每个cell barcode 关联的空间条形码的坐标密度,统计在网格bin上的UMI分布;以最大密度bin为细胞中心,并结合核心-次中心比值校验唯一性。
- 输出
cell_location.tsv.gz记录细胞在芯片坐标系中的位置,实现表达矩阵的空间锚定与可视化叠加。
以上详细内容可以查看SeekSpace Tools
SeekSpace Tools生成的结果文件可直接用在云平台进行后续分析。
初始阶段
【新建流程】创建单细胞分析流程。项目开始一般选择样本进行大群分析,后续可选择注释好的细胞类型进行亚群分析。
填写“流程名称”及“流程描述”,用于后续查找和了解流程信息。
【选择数据】选择待分析的样本,可选择已经整合好的多样本数据,也可选多个单样本进行过滤整合。
NOTE
数据详细来源可查看《我的数据》。
- 填写信息,选择待分析的数据,可以选择【开始分析】手动进行过滤整合和聚类,也可以选择【基础分析】、【基础分析+细胞注释】、【基础分析+细胞注释+差异富集】或【基础分析+细胞注释+差异富集+画图工具】快速进行自动分析。
过滤
- 【开始分析】后的“图表数据”会统计样本UMI、线粒体占比及基因表达情况,简要展示各样本质量信息。
NOTE
指标释义(scRNA-seq):
- nCount_RNA:每细胞 UMI 总数,反映测序深度与转录本丰度
- nFeature_RNA:每细胞检出的基因数,反映表达复杂度
- 线粒体比例(mito):线粒体基因 UMI 占比,偏高常与凋亡/损伤相关(高代谢组织可适度放宽)
- 展开按钮可查看默认参数,用户可参考已发表单细胞文章方法中的过滤参数进行【过滤】。
TIP
如果选择的是已经整合的数据,点击【过滤】会提示是否需要跳过过滤整合步骤。如果不需要调整参数可跳过进行后续分析,如需调整过滤阈值则取消跳过,重新进行过滤整合。
IMPORTANT
过滤策略建议:
- 使用 MAD 动态阈值过滤 mt% 或以 10%-20% 作为参考上限;
- nCount_RNA/nFeature_RNA 以分位数或箱线图上界识别极端异常;
- 多样本时可按样本细胞数做均衡抽样,避免样本量主导聚类;
- 高代谢组织(心/肾/肝等)与免疫/粒细胞丰富样本需适当放宽阈值,避免误删真实细胞。
【过滤】后可查看各样本过滤后质量信息,同时可对单样本进行个性化调整,保证整体样本质量一致。
CAUTION
避免机械性依赖“拉高细胞数”的操作(如仅凭经验值强行放宽下限)。若瀑布图拐点不清、背景高或低 UMI 群占比异常,强行回收会降低下游稳定性。 双胞(doublet)排查思路:
- 识别 nCount_RNA/nFeature_RNA 上尾细胞(分布右侧极端高值);
- 是否存在互斥 marker 共表达、UMAP 两团之间的“桥状”细胞;
- 肿瘤项目可结合 CNV 辅助判定;
- 谨慎过滤:建议多证据一致时再剔除,并在剔除后重做整合与聚类验证一致性。
整合
质控合格的数据进行【整合】,目前提供四种整合方法,其中CCA、Harmony和RPCA会对多个样本进行批次矫正。
【整合】后会展示样本整合情况,可调整整合参数重新整合。建议用户尝试多种整合方法,选择更合适的方法进行后续分析。
TIP
整合方法选择:
- 批次效应较弱:merge 直接合并,避免过度校正;
- 中等批次:CCA/RPCA;
- 批次显著或异构明显:Harmony 更稳健。
TIP
效果评估三准则:
- 批次混合度:同一细胞类型在 UMAP/TSNE 中跨样本均匀混合;
- 生物信号保留:经典 marker 梯度与分群边界清晰,差异/富集结果符合预期;
- 过度/欠校正告警:过度校正会抹平差异,欠校正会出现“按批次聚类”。
聚类
【整合】确认后进行【聚类】,可新建选择多个分辨率进行聚类,分辨率越大分群数量越多。后续模块也可新增分辨率进行聚类。
TIP
聚类调参与排错:
- 拐点法:以 PCA 肘部拐点作为 dims 起点,细胞量越大适当增加;
- 子集重聚类:如 T 细胞等大类单独重聚类,放大类内异质性。 dims 过低会遗漏关键异质性,过高易过聚类与放大噪音。请结合拐点与重现性优选。
查看更多内容(展开/收起)
空间聚类与RNA聚类整合(非必需)
Banksy 与 RNA 聚类的关系
- RNA 聚类仅基于基因表达,擅长区分细胞类型/状态,但可能忽略空间连续性与微环境线索。
- 空间聚类引入邻域依赖与空间梯度,擅长识别组织功能域与边界,但可能牺牲部分表达分辨率。
- Banksy 在表达特征上叠加邻域统计特征(如邻域均值/梯度),通过可调权重进行联合聚类,用于“修正/细化”RNA 聚类边界、合并空间上不连续的伪簇,或揭示被 RNA 聚类忽略的空间功能域。
整合思路(基于云平台工作流)
- 云平台 RNA 聚类基线 → 导入 Banksy CSV → 一致性评估与差异富集
- 在云平台完成标准 RNA 聚类,得到表达层的基线标签。
- 获取高级分析 Banksy 结果 CSV(如
XXX_banksy_colData.csv),通过“上传合并meta”并入流程 meta(须包含barcode,列名避免与流程内置字段冲突)。 - 在平台上对比 RNA 基线与 Banksy 簇/空间域的一致性(NMI/ARI、空间连通性/断裂率、域内表达同质性),并以 Banksy 或一致性优化后的标签为分组做差异表达、富集(GO/KEGG/通路)与空间可视化。
TIP
(为何与如何整合):
- 基于“自身表达 + 空间微环境表达”联合建模:表达决定“它是什么”,空间决定“它在哪里、它做什么”。
- 能识别具有特定空间位置的细胞亚型:揭示仅在特定区域出现的功能性细分亚群。
- 利用邻居信息作为佐证提高置信度:邻域一致的表达模式可帮助合并伪簇、削弱噪声。
- 聚类结果更自然地对齐空间区域:更好地对应组织学结构与形态学边界。
Banksy 并不是替代传统单细胞注释的方法,而是一个强大的增强工具:在既有的 RNA 基线之上注入关键的空间维度,把对细胞身份的理解从“它是什么”推进到“它在哪里、它做什么”的更高层次。
- 云平台 RNA 聚类基线 → 导入 Banksy CSV → 一致性评估与差异富集
关键参数与实践建议(云平台参数面板)
- algo:聚类方法,支持
leiden、louvain、kmeans、mclust。 - 分辨率/簇数:
leiden/louvain用resolution(越大簇越多,结合空间连通性与 marker 解释性微调)。kmeans用kmeans.centers;mclust用mclust.G(均为簇数)。
- lambda:表达与空间位置的权重;
0表示不引入空间,常用0.1–0.3,结构清晰可略升,噪声大/稀疏样本可适度降低避免过度平滑。 - 主成分数量:用于构建特征空间的 PC 数,默认
30,数据规模大或异质性强可适度上调。
- algo:聚类方法,支持
TIP
调参建议:
- 先在 RNA 聚类上确定稳定的表达基线,再在小范围内网格化尝试
lambda与分辨率(或簇数)组合。 - 优先选择兼顾较高 NMI/ARI 与更低空间断裂率的结果;当 NMI 略降但空间域更连续且 marker 更一致时,倾向选择 Banksy 标签。
TIP
实践建议:先固定 RNA 分辨率获得稳定的表达基线,再调节 lambda 与 k_geom 观察空间连通性与 NMI 的变化;当 NMI 下降但空间断裂率显著降低且 marker 呈现更清晰的空间域时,优先选择 Banksy 标签。
- 一致性、解释性与交付
- 一致性:报告 NMI/ARI、空间断裂率、域内表达同质性与空间自相关(如 Moran's I)。
- 可解释性:建立 Banksy 簇与 RNA 类型的映射,核验域内 marker、组织学区域与已知结构的一致性。
- 结果交付:
- 输出“RNA 类型 × 空间域”的交叉表与并排可视化(UMAP 与空间坐标)。
- 基于差异与富集结果,附上显著通路与代表基因的空间热图,阐明空间功能差异。
参考与延伸阅读
Banksy 资料:
分析完成
【聚类】后无需调整则点击【完成】,跳转“细胞注释”模块,正式开始进行单细胞相关分析。该步会耗费一定时间,请用户耐心等待。