inferCNV拷贝数变异分析文档
前言
TIP
inferCNV是单细胞转录组领域用于推断肿瘤细胞拷贝数变异(Copy Number Variation, CNV)的主流生物信息学工具。它通过分析基因表达数据,识别出单个细胞或细胞群中可能存在的染色体片段扩增或缺失,从而有效区分肿瘤细胞与正常细胞,并揭示肿瘤内部的克隆异质性。
在肿瘤研究中,基因组的不稳定性是其核心特征之一,而CNV是导致这种不稳定性的重要原因。通过在单细胞水平上解析CNV,研究者可以:
- 精确识别恶性细胞:将携带大量CNV的肿瘤细胞与混杂在肿瘤组织中的正常免疫细胞、基质细胞等区分开。
- 探究肿瘤异质性:识别具有不同CNV特征的肿瘤亚克隆,了解肿瘤的演进过程。
- 关联基因型与表型:将特定的CNV事件与细胞的功能状态、耐药性等表型联系起来。
本篇文档旨在为研究者提供一份详尽的inferCNV技术指南,内容涵盖其基本原理、在SeekSoulOnline云平台上的操作方法、结果解读及实战案例。
inferCNV理论基础
核心原理
inferCNV的核心思想是:在一个混合的细胞群体中,如果能以一组基因组稳定的细胞作为“正常”参照,就可以通过比较其他细胞的基因表达水平,来推断其是否存在染色体片段的拷贝数变化。
它基于一个关键假设:特定染色体区域的基因平均表达水平与其拷贝数成正比。如果一个区域发生扩增,该区域内基因的总体表达水平会上升;反之,如果发生缺失,则总体水平会下降。
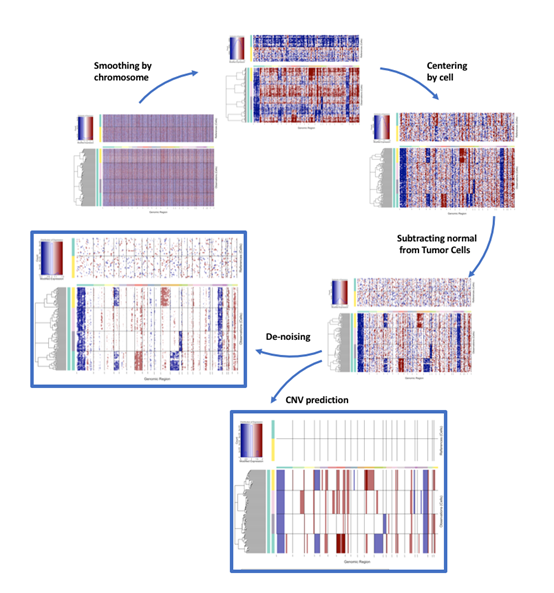
分析流程概览
- 定义细胞组:将细胞分为两组:待分析的“观测组”(Observation)和作为基准的“参照组”(Reference)。参照组通常是已知的正常细胞,如T细胞、B细胞或成纤维细胞。
- 计算相对表达:计算观测组中每个细胞、每个基因相对于参照组平均表达的log2(fold change)。
- 平滑噪声:为了消除单个基因表达的随机波动,inferCNV会沿着染色体,使用移动窗口(如101个基因)对基因的相对表达值进行平滑处理。这一步能有效凸显由大片段CNV引起的持续性表达变化。
- 中心化和去噪:对数据进行中心化,使基准线回到0。同时,可以应用特定算法去除背景噪声。
- 结果可视化:最终通过热图展示CNV谱。热图的行是细胞,列是按染色体位置排序的基因。红色代表拷贝数增加,蓝色代表拷贝数减少。
云平台操作指南
在云平台上,inferCNV分析流程被设计得直观易用,无需编写代码。
参数详解
| 界面参数 | 说明 |
|---|---|
| 任务名称 | 本次分析的任务名称,需以英文字母开头,可包含英文字母、数字、下划线和中文。 |
| 物种 | 选择该分析流程数据对应的物种名称,目前支持人、小鼠和大鼠。 |
| 分组因子 | 选择要分析的细胞类型或者聚类对应的标签。例如,若想对注释好的细胞类型进行分析,则选择其对应的标签(如CellAnnotation),并与下方的“参考/分析细胞类型”配合使用。 |
| 参考细胞类型 | (核心参数) 多选,选择作为正常参照的细胞类型,通常为免疫细胞或正常组织的上皮细胞。可选择分组因子中的所有水平。 |
| 分析细胞类型 | (核心参数) 多选,选择待分析的细胞类型,通常为癌变组织的上皮细胞。可选择分组因子中除参考细胞类型外的所有水平。 |
| 基因最小平均表达量 | 基因的平均表达量阈值,范围为(0,1],默认是0.1。 |
| 热图图注 | 按照所选标签为热图添加分组注释。 |
| downsample | 是否对细胞进行降采样(抽取部分细胞)进行分析。 |
| downsample_num | 若启用降采样,此处设置抽取的细胞数量。 |
| 备注 | 自定义备注信息。 |
| 附加参数 | 是否修改默认高级参数,默认不修改。 |
| cluster_by_groups | 当k_obs_groups为FALSE时,可在此自定义分析细胞类型的最终聚类分群数目。 |
| k_obs_groups | 选择TRUE时,会先对每个分析细胞类型各自聚类,再进行总的层次聚类。选择FALSE时,则对所有分析细胞类型进行整体聚类。 |
| hclust_method | 细胞层次聚类的方法,包括ward.D、ward.D2(默认)、single、complete等。 |
| denoise | 是否进行降噪处理,默认开启。 |
| window_length | 基因表达平滑窗口的大小(基因个数),需要是奇数。 |
TIP
参考细胞的选择至关重要。错误地将肿瘤细胞选为参考,或将携带CNV的细胞混入参考集,都会严重影响结果的准确性。通常建议选择与肿瘤细胞来源不同胚层的细胞作为参考(例如,对于上皮来源的肿瘤,选择免疫细胞或基质细胞)。
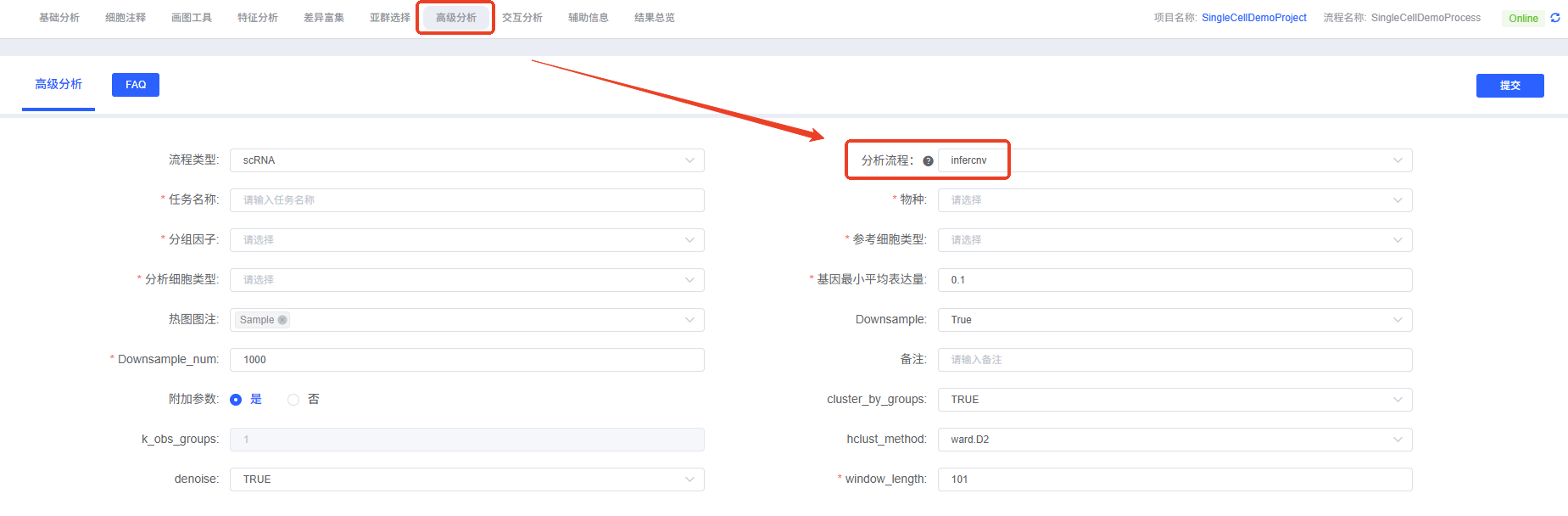
操作流程
- 进入分析模块:在云平台导航至“高级分析”模块,选择“infercnv”。
- 创建新任务:为您的分析任务命名,并选择要分析的样本或项目。
- 配置参数:根据上述指南,选择要分析的细胞类型、分组信息等。
- 提交任务:确认参数无误后,点击“提交”按钮,等待分析完成。
- 下载与查看:分析结束后,在任务列表中下载并查看生成的分析报告和结果文件。
结果解读
inferCNV的分析报告包含丰富的图表和数据文件,以下是对核心结果的详细解读。
CNV分布总览热图
这是最核心和最直观的结果,展示了所有细胞在全基因组范围内的CNV模式。

- 图表解读:
- 行:代表单个细胞,通常会按照细胞类型或CNV模式进行聚类。
- 列:代表按染色体位置(从1号到X/Y号)排序的基因。
- 颜色:红色表示基因表达相对于参考细胞上调,推测为拷贝数增加;蓝色表示基因表达下调,推测为拷贝数减少。
- 上下分区:热图通常分为上下两部分,上方为参考细胞(Reference),下方为待分析细胞(Observation)。
- 分析要点:
- 观察参考细胞区域是否基本为中性色,这表明参考细胞选择得当。
- 观察待分析细胞中是否存在大片段的、连续的红色或蓝色区域,这些就是推断出的CNV事件。
- 比较不同细胞或细胞群的CNV模式,可以识别出具有不同基因组变异特征的肿瘤亚克隆。
按细胞类型/分组展示的CNV热图
此图更聚焦于待分析的细胞,并按预先注释的细胞类型进行分组展示。
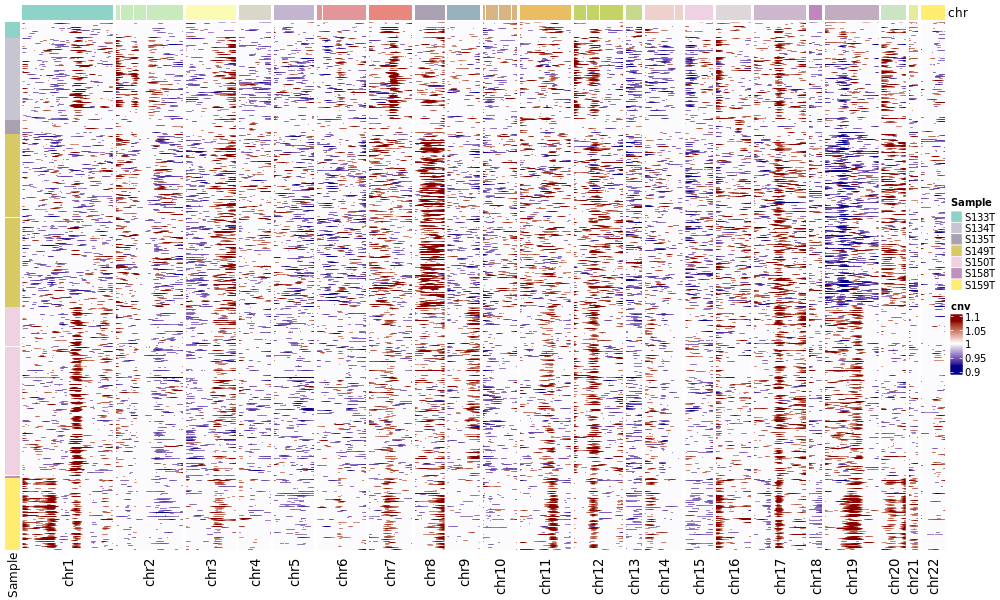
- 图表解读:此图只展示待分析细胞(Observation),并根据您提供的细胞类型注释对细胞(行)进行排序和颜色标记。
- 分析要点:
- 确认CNV事件是否主要富集在预期的肿瘤细胞类型中。
- 评估肿瘤内部的异质性。例如,某些肿瘤细胞亚群是否携带了其他亚群没有的特定CNV事件。
CNV Score
为了量化每个细胞的CNV水平,inferCNV会计算一个CNV Score,反映了细胞基因组中CNV信号的整体强度。
CNV Score降维图
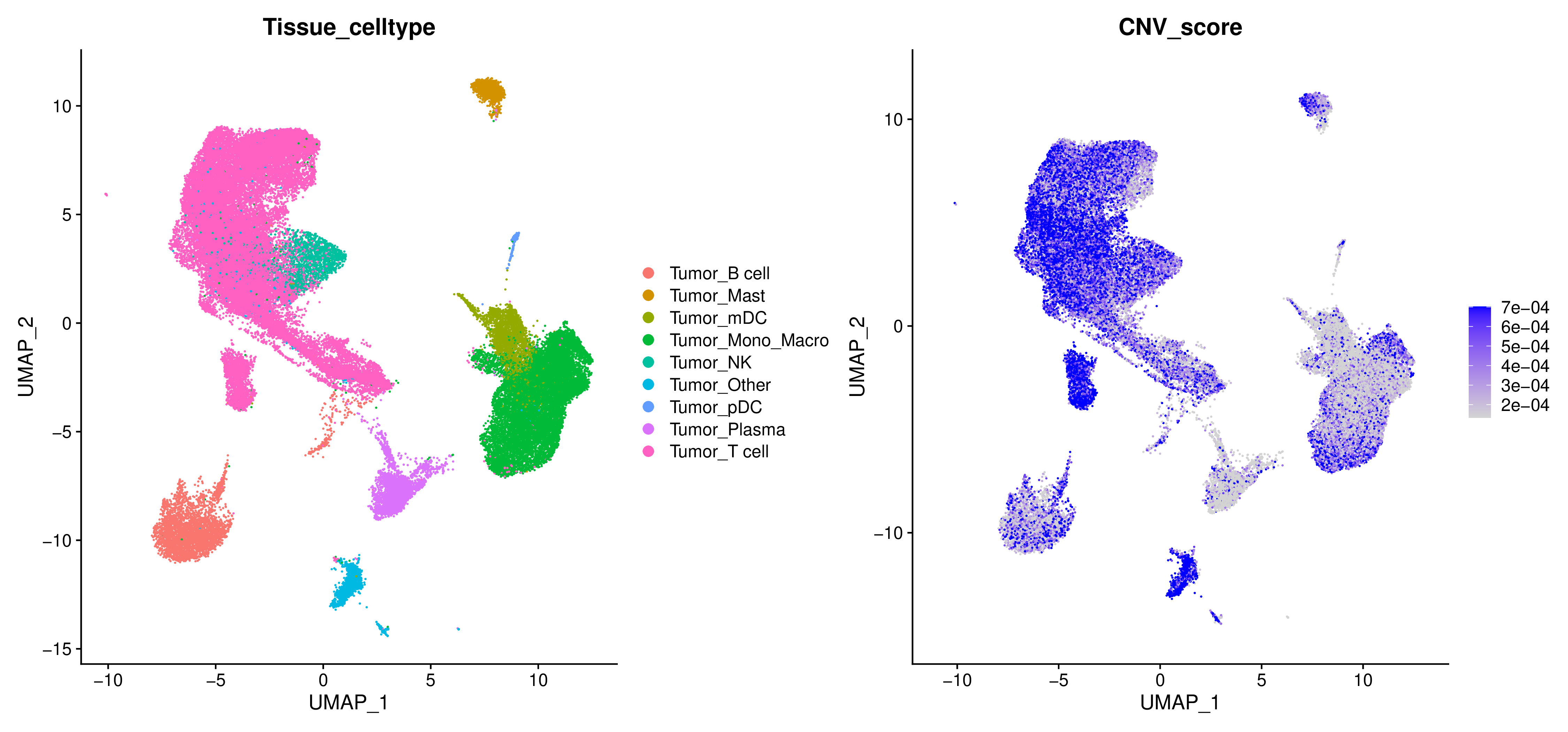
- 图表解读:将CNV Score投影到UMAP或t-SNE降维图上。颜色越深,代表CNV Score越高,即该细胞的基因组越不稳定。
- 分析要点:直观地识别出哪些细胞聚类是潜在的肿瘤细胞(通常为高CNV Score的区域)。
CNV Score小提琴图
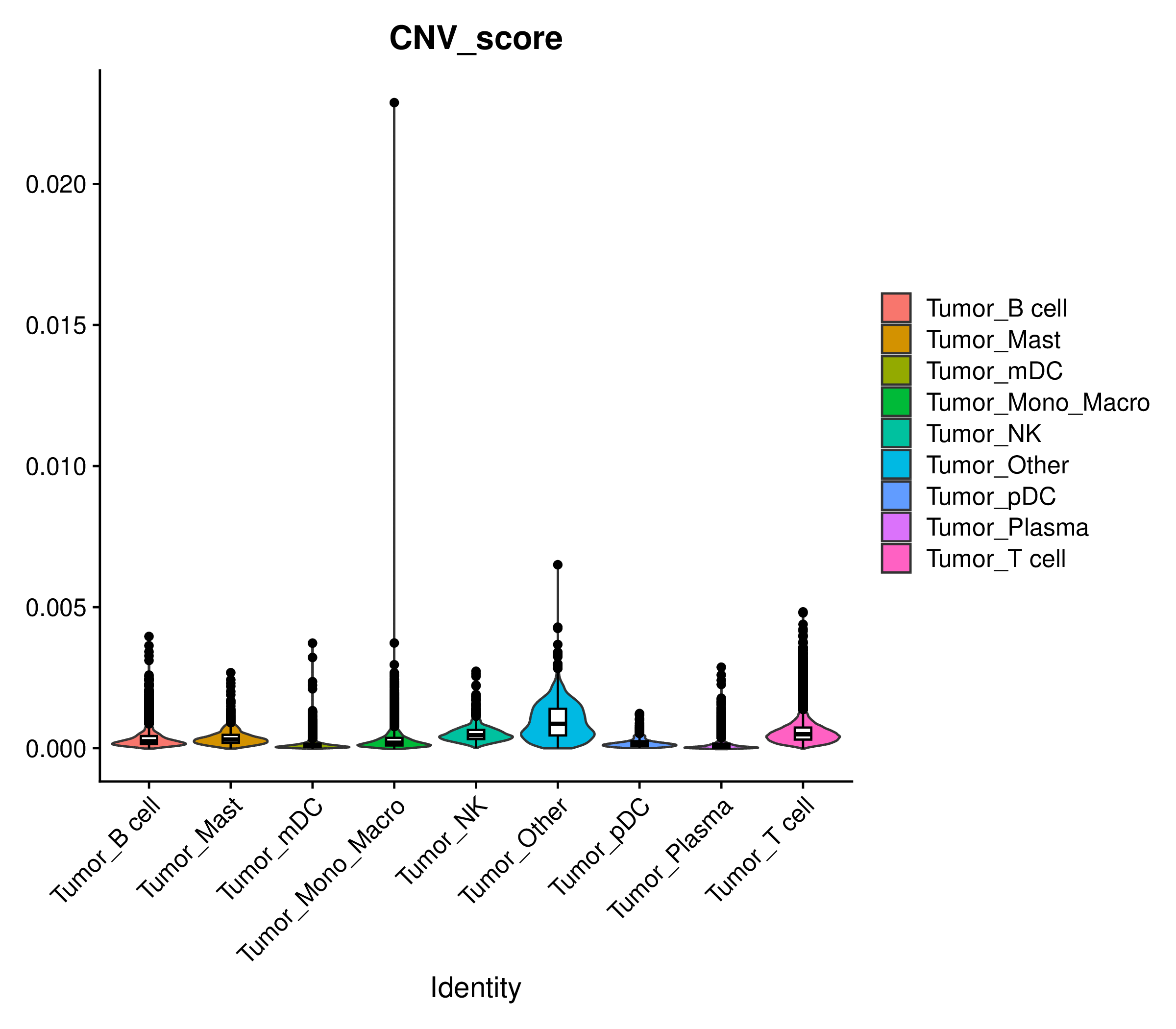
- 图表解读:按细胞类型展示每个细胞群的CNV Score分布。
- 分析要点:定量比较不同细胞类型的CNV水平。通常,肿瘤细胞群的CNV Score会显著高于正常细胞群。
应用案例
案例一:肺腺癌(文献解读)
研究背景
在2021年发表于Science Advances的一项研究中,研究人员利用单细胞转录组技术揭示了肺腺癌的肿瘤异质性。其中,inferCNV被用于区分肿瘤细胞和非肿瘤细胞,并验证其与全外显子测序(WES)结果的一致性。
分析结果
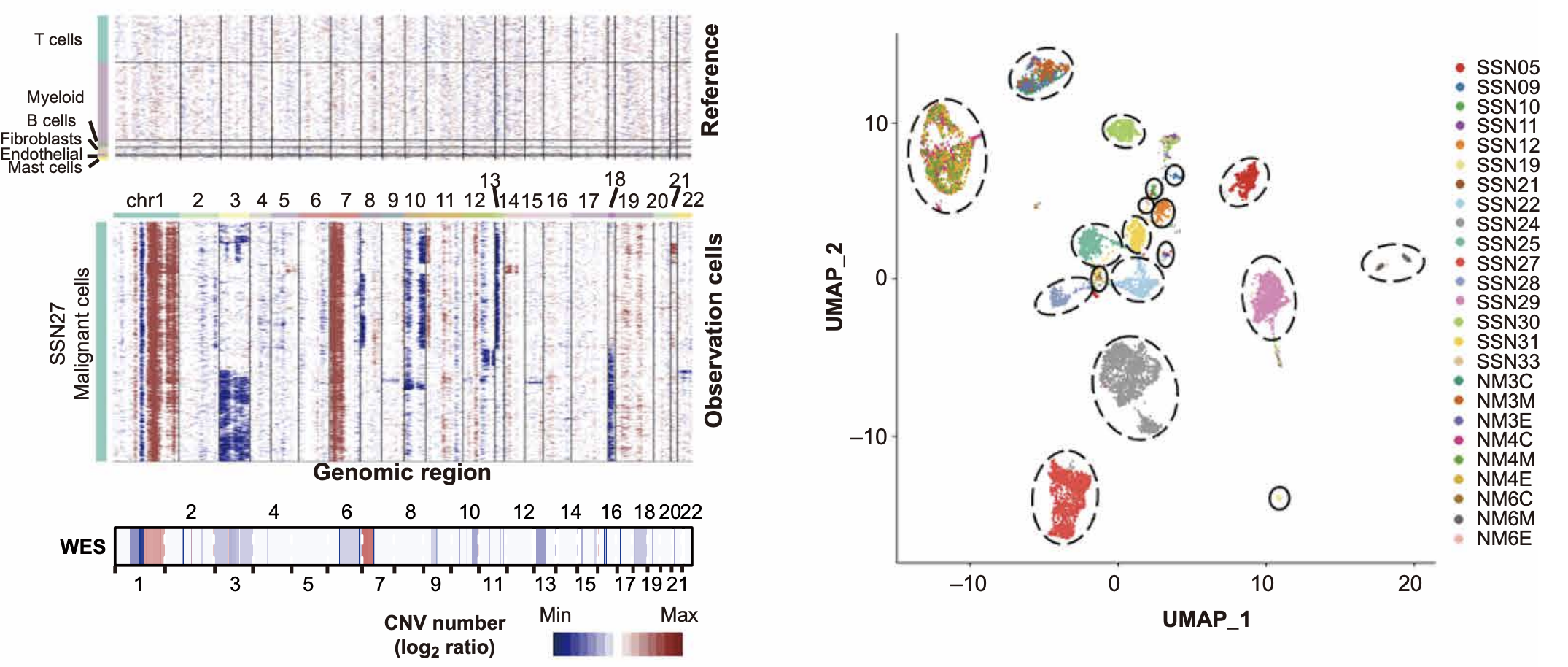
- 左图:展示了单个样本的inferCNV结果。以非恶性细胞(免疫细胞和基质细胞)为参考(上图),可以清晰地看到恶性细胞(中图)中存在大量与WES检测到的CNV模式(下图)高度一致的拷贝数变异事件。
- 右图:UMAP图显示,来自不同患者的肿瘤细胞在很大程度上分开聚集,表明肿瘤间存在高度的异质性。
结论
该案例表明,inferCNV能够:
- 有效区分肿瘤与非肿瘤细胞。
- 揭示肿瘤内部异质性。
- 结果可靠,与金标准(如WES)具有良好的一致性。
注意事项
1. 参照组的选择至关重要:inferCNV分析的准确性极度依赖于一个高质量的、确定为“正常”二倍体的参照细胞群。如果参照组选择错误(例如,混入了肿瘤细胞,或选择的细胞类型本身存在生理性的基因表达差异),将会导致整个分析结果出现系统性偏差,甚至得出完全错误的结论。
2. 信噪比问题:单细胞转录组数据本身具有高噪音和高稀疏性的特点。inferCNV通过在染色体上平滑基因表达来增强信号,但这依然可能受到技术噪音的影响。对于CNV信号微弱的肿瘤,结果可能不明显或不可靠。
3. HMM模式的应用:当您需要更明确的CNV状态分类(如“扩增”、“缺失”、“中性”)而不仅仅是相对表达的热图时,可以考虑使用HMM(隐马尔可夫模型)预测模式。这有助于更客观地定义CNV区域,但对数据质量要求更高。
4. 结果不是绝对拷贝数:inferCNV展示的是相对于参照组的基因表达水平变化,它反映的是相对拷贝数,而不是绝对拷贝数(如2拷贝、3拷贝)。
常见问题解答 (FAQ)
Q1: 为什么我的CNV热图上,所有细胞(包括参考细胞)看起来都有CNV?
A1: 这是一个常见现象,通常是由技术噪音或不完美的参考细胞引起的。inferCNV的结果是相对的,关键在于比较“观测组”和“参照组”的模式差异。如果观测组显示出比参照组更强、更一致的大片段CNV模式,那么这些信号通常是可靠的。此外,可以尝试更换参考细胞类型或启用denoise模式来优化结果。
Q2: inferCNV能否区分杂合性缺失(loss of heterozygosity, LOH)?
A2: 不能。inferCNV是基于转录组数据的,它只能推断总的拷贝数变化(增加或减少),无法区分等位基因。LOH的精确鉴定需要基因组测序数据。
Q3: 我的分析失败了,可能是什么原因?
A3: 常见原因包括:1) 参考细胞选择错误:您在参数中选择的“参考细胞类型”在您的细胞注释数据中不存在或名称不匹配。2) 基因数量过少:经过“基因最小平均表达量”过滤后,剩余的基因太少,无法进行有效的平滑分析。3) 物种与基因ID不匹配:选择的物种(如human)与您表达矩阵中的基因ID(如小鼠基因)不符。
参考文献
- Patel, A. P., et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 344(6190), 1396-1401.
- Tirosh, I., et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science, 352(6282), 189-196.
- Xing, X., et al. (2021). Decoding the multicellular ecosystem of lung adenocarcinoma manifested as pulmonary subsolid nodules by single-cell RNA sequencing. Science Advances, 7(5), eabd9738.
- Kumar, M., et al. (2020). Single-cell analysis of copy-number alterations in serous ovarian cancer reveals substantial heterogeneity in both low- and high-grade tumors. Cell Cycle, 19(23), 3154-3166.
- Wu, F., et al. (2021). Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nature Communications, 12(1), 2540.
- inferCNV Wiki