Monocle2拟时序分析文档
前言
TIP
Monocle2是单细胞转录组学领域应用最广泛的拟时序分析工具之一。它能够帮助研究者重建细胞在发育、分化或疾病进展等动态生物学过程中的连续轨迹,揭示细胞命运决定的关键调控机制。
在单细胞研究中,我们观察到的细胞状态往往是异质且异步的。例如,在同一时间点捕获的细胞群体中,有的细胞可能处于分化早期,而另一些则已接近终末状态。拟时序(Pseudotime)分析通过生物信息学算法,将这些静态的细胞快照按照其内在的发育进程进行排序,从而构建出一条或多条连续的细胞分化轨迹。
Monocle2的核心功能
- 细胞轨迹构建:重建细胞从一种状态到另一种状态的转变路径,尤其适用于分析包含多个分支(即细胞命运决定点)的复杂过程。
- 基因动态变化分析:识别在拟时序轨迹上表达模式发生显著变化的基因,找到驱动细胞状态转变的核心调控因子。
- 细胞状态定义:基于轨迹将细胞划分为不同的发育阶段(State),并对这些阶段进行功能表征。
本篇文档旨在为单细胞研究者提供一份详尽的Monocle2技术指南,内容涵盖其基本原理、在SeekSoulOnline云平台上的操作方法、结果解读、实战案例及常见问题,帮助您快速掌握并应用该工具。
Monocle2理论基础
核心原理
Monocle2的核心思想是:如果能找到一个能够代表细胞动态发展过程的基因集,就可以根据这些基因在每个细胞中的表达模式,将细胞排列在一个低维空间中,从而重建出它们的动态变化轨迹。 这个过程可以概括为三步:
Monocle2的轨迹推断主要依赖于**DDRTree(Discriminative Dimensionality Reduction via learning a Tree)**算法。其核心思想可以分解为三个主要步骤:
选择排序基因 (Ordering Genes):首先,需要挑选出一组在不同细胞状态间差异表达的基因。这些基因的表达变化能够代表细胞所处的生物学进程。Monocle2提供多种选择策略,最常用的是选择在细胞间表达变异程度高的基因。
降维 (Dimension Reduction):利用选出的排序基因,Monocle2采用DDRTree算法进行降维。该算法不仅能降低数据维度,还能同时学习到一个能够描述数据内在结构的“主图”(Principal Graph),这个图是后续构建轨迹的基础。
细胞排序 (Placing Cells on the Trajectory):将每个细胞投射到学习到的主图上,从而确定其在轨迹上的位置,并计算出拟时序值。算法会找到图中最长的一条路径作为“主干”,并以此为基础识别可能的分支点和不同的细胞命运终点。
关键算法:DDRTree
DDRTree是Monocle2与Monocle早期版本及其他拟时序分析工具(如TSCAN)的关键区别之一。它是一种“反向图嵌入”(Reversed Graph Embedding)的方法,其优势在于:
- 同时降维和聚类:在降维的同时,将细胞聚类到图的节点上。
- 鲁棒的轨迹构建:能够有效地学习到复杂的生物学轨迹,特别是包含多个分支点的轨迹。
- 结果稳定性:相比于基于最小生成树(MST)的方法,DDRTree对数据中的噪声和异常值不那么敏感。
TIP
理解DDRTree是掌握Monocle2的关键。它通过构建一个“树状”的低维结构来拟合高维的基因表达数据,这棵“树”的树干和树枝就构成了我们最终看到的细胞分化轨迹。
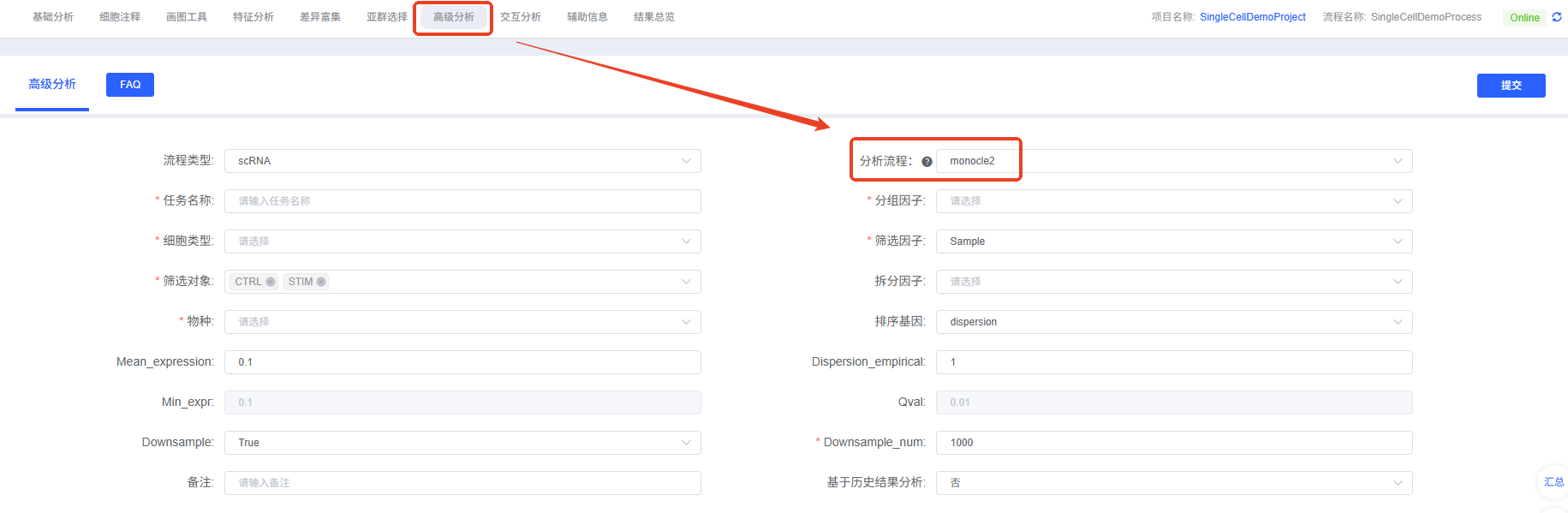
云平台操作指南
在云平台上,Monocle2分析流程被设计得直观易用。您无需编写代码,只需通过参数配置界面即可完成分析。
分析前的准备
TIP
Monocle2分析的成功与否,很大程度上取决于输入数据的质量和生物学问题的合理性。在开始分析前,请务必确认:
- 数据已完成预处理:您的单细胞数据已经过标准的质控、降维、聚类和细胞类型注释。
- 选择了合适的细胞亚群:拟时序分析应在具有潜在分化或转变关系的细胞亚群中进行。例如,分析不同亚型的T细胞,或不同分化阶段的祖细胞。将生物学上毫无关联的细胞(如T细胞和上皮细胞)放在一起进行分析是没有意义的。
- 明确生物学起点:虽然Monocle2可以自动推断轨迹的起点,但如果您的研究中有明确的生物学起点(如干细胞、初始T细胞等),了解这一点将有助于后续的结果解读。
参数详解
下表详细列出了云平台Monocle2分析模块的主要参数及其说明。
| 界面参数 | 说明 |
|---|---|
| 任务名称 | 本次分析的任务名称,需以英文字母开头。 |
| 分组因子 | 选择要分析的细胞类型或聚类对应的标签,例如celltype。 |
| 细胞类型 | 多选,选择要纳入分析的具体细胞类型或聚类。 |
| 筛选因子 | 选择用于筛选样本或分组的标签,例如Sample。 |
| 筛选对象 | 多选,选择要保留的样本或分组。 |
| 拆分因子 | 多选,绘图时用于拆分不同组别的标签,如Group或Sample。 |
| 物种 | 选择您的数据对应的物种。 |
| 排序基因 | 选择用于构建轨迹的基因集策略。 |
a. dispersion | 高变基因(默认)。基于基因表达的离散度选择,无需先验知识。 |
b. differential | 差异基因。基于不同细胞群间的差异表达基因进行选择。 |
c. genelist | 自定义基因。上传您自己定义的基因列表。 |
| Mean_expression | dispersion模式参数,高变基因的平均表达阈值。 |
| Dispersion_empirical | dispersion模式参数,经验离散度阈值。 |
| Min_expr | differential模式参数,差异基因的最低表达阈值。 |
| Qval | differential模式参数,差异基因的q值(FDR)阈值。 |
| 自定义差异基因集 | genelist模式参数,点击【模版下载】,按照模版格式上传基因列表文件。 |
| Downsample | 是否对大数据集进行随机下采样。 |
| Downsample_num | 每个细胞亚群下采样保留的细胞数目。 |
| 基于历史结果分析 | 选择“修改root”,可基于已完成的分析任务重新指定起点。 |
| 历史任务名称 | 选择一个已成功的Monocle2任务。 |
| root | 在历史任务结果中,选择一个新的细胞状态(State)作为起点。 |
| 备注 | 自定义备注信息。 |
重要注意事项
TIP
- 大数据集处理:当细胞总数超过数万时,若
Downsample参数设置为False,分析可能会因内存不足而失败。强烈建议开启Downsample进行分析。 - Metadata规范:请确保RDS文件中的metadata列名和内容不包含中文或特殊字符(如
&),否则可能导致流程错误。
操作流程
- 进入分析模块:在云平台导航至“高级分析”模块,选择“monocle2”。
- 创建新任务:为您的分析任务命名,并选择要分析的样本或项目。
- 配置参数:根据上述指南,选择要分析的细胞类型、分组信息等。
- 提交任务:确认参数无误后,点击“提交”按钮,等待分析完成。
- 下载与查看:分析结束后,在任务列表中下载并查看生成的分析报告和结果文件。
结果解读
Monocle2的分析报告包含丰富的图表和数据文件,以下是对核心结果的详细解读。
5.1 细胞轨迹总览图
这是Monocle2分析最核心和最直观的结果。它将所有细胞展示在一个二维空间中,并用线条连接成树状结构,代表细胞的发育轨迹。
5.1.1 Pseudotime分布图
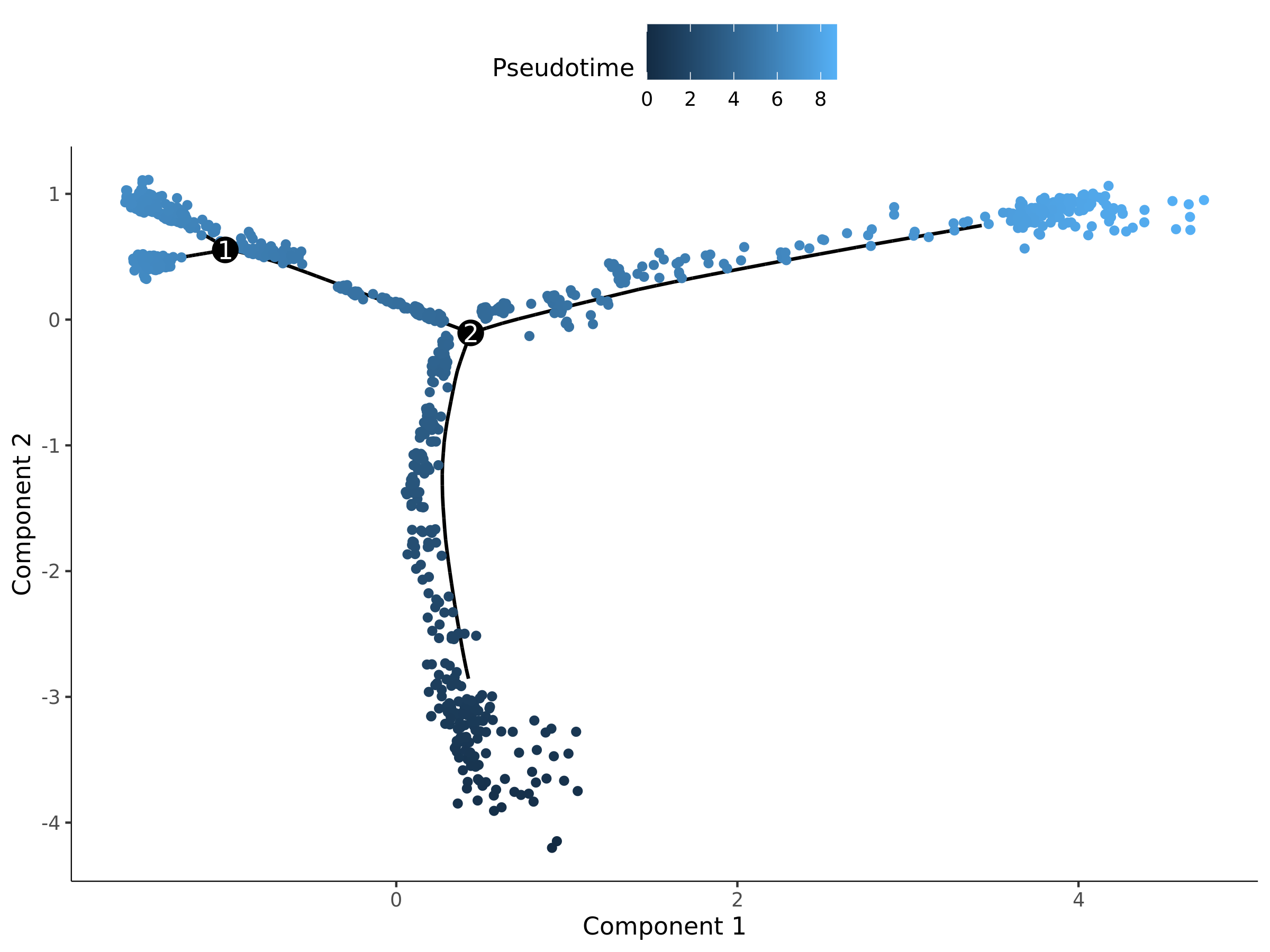
- 图表解读:每个点代表一个细胞。颜色代表该细胞的拟时序值,通常从蓝色(早期)到黄色(晚期)渐变。拟时序值越小,代表细胞越接近发育的起点;值越大,则越接近终点。
- 分析要点:
- 观察轨迹的整体形状:是线性的单向分化,还是包含分支点的多命运决策?
- 识别起点和终点:颜色最深的区域通常是轨迹的起点。
5.1.2 State分布图
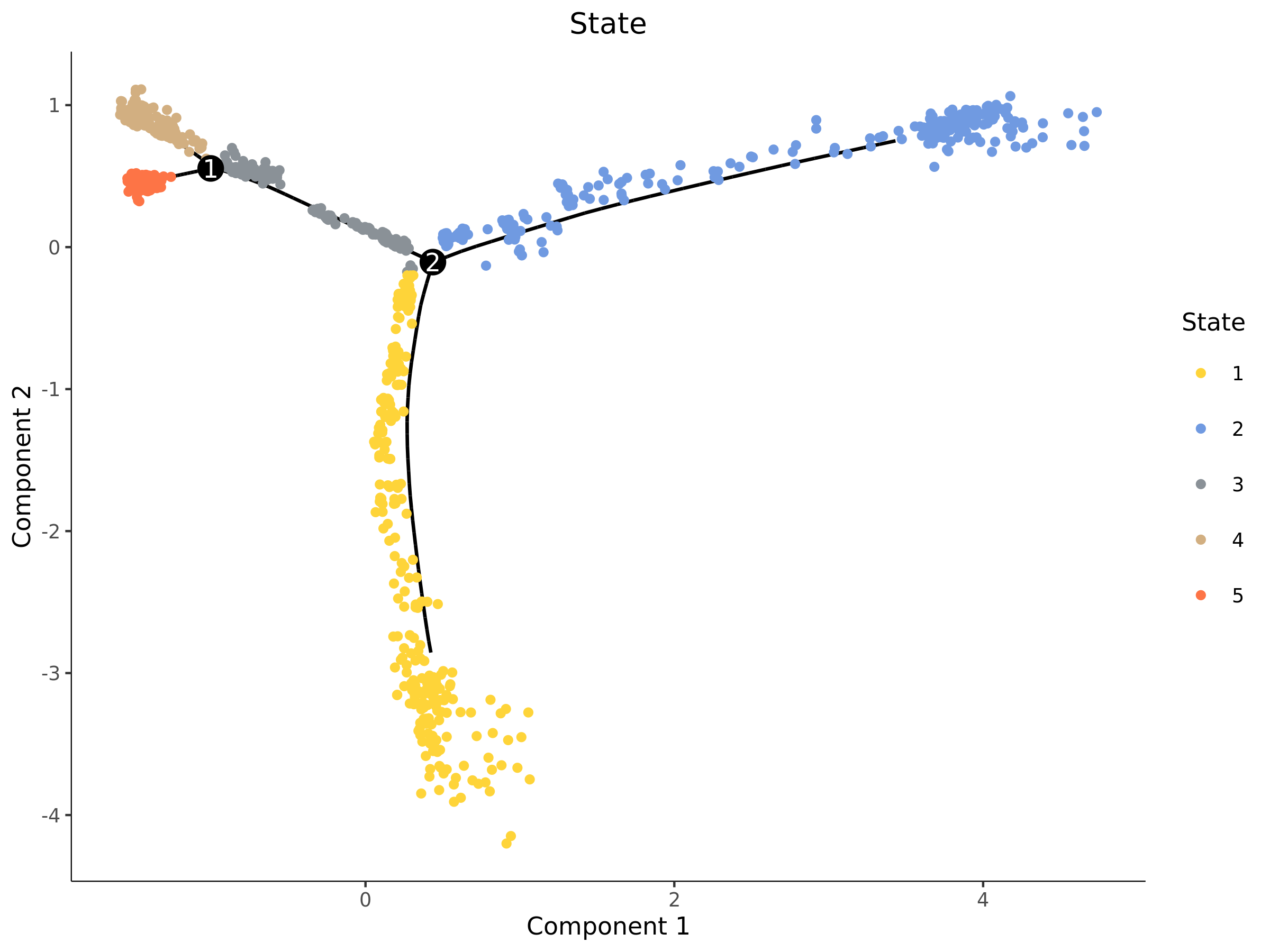
- 图表解读:Monocle2会将轨迹上的细胞划分为不同的“状态”(State),可以理解为细胞在发育途中的不同阶段。图中用不同的颜色表示不同的State。
- 分析要点:
- State的分布与轨迹分支密切相关。一个分支点(Branch Point)通常连接着一个“前体”State和两个或多个“子代”State。
- 结合后续的基因表达分析,可以探究每个State的功能特征。
5.1.3 细胞群(celltype)分布图
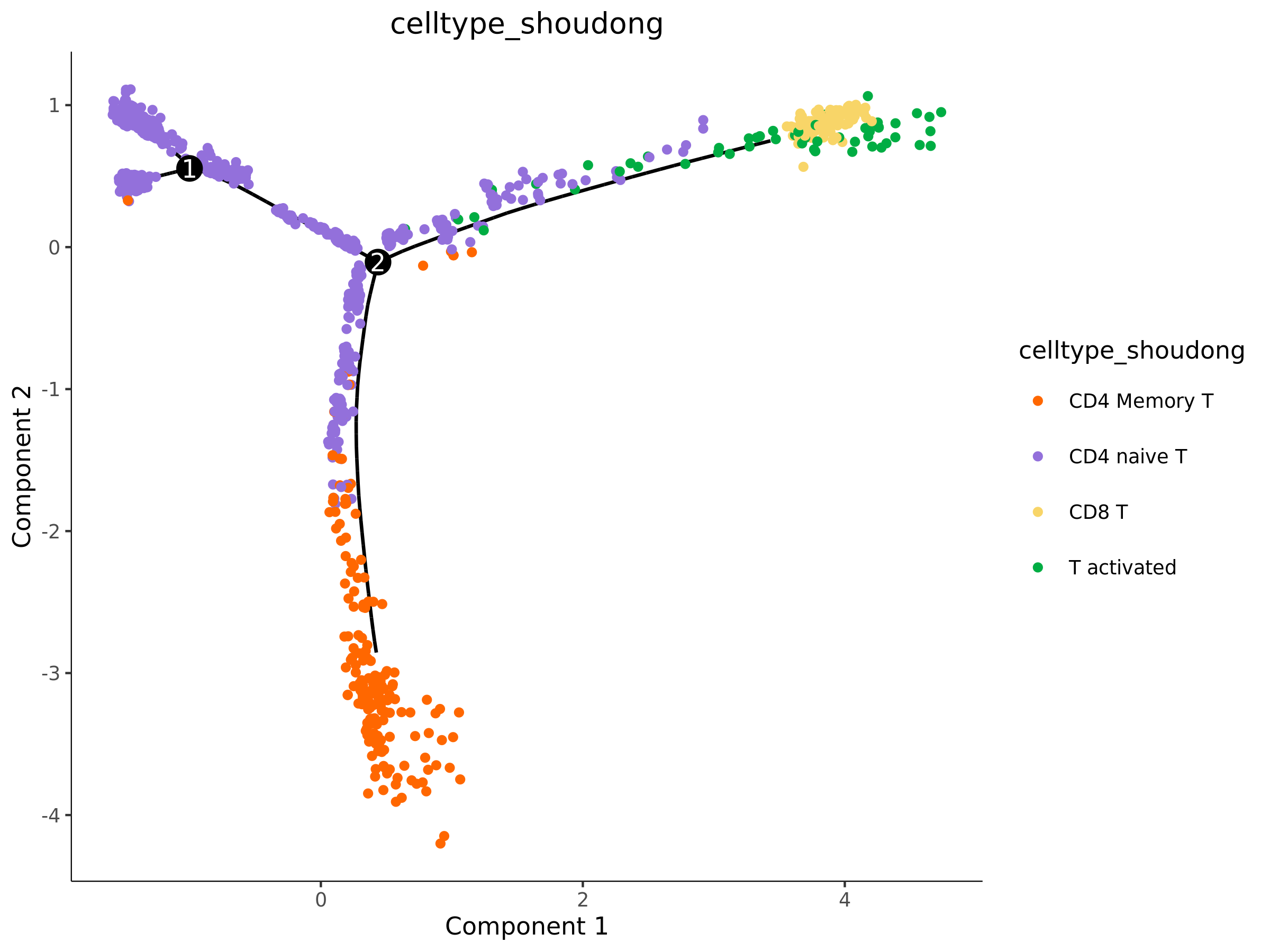
- 图表解读:这是至关重要的一张图。它将您预先注释的细胞类型(Cell Type)映射到轨迹上,用不同颜色表示。
- 分析要点:
- 验证轨迹的生物学意义:检查细胞类型的分布是否符合已知的生物学过程。例如,干细胞是否位于起点?终末分化的细胞是否位于轨迹的末端?
- 确定分化方向:通过观察细胞类型的连续排布,可以推断出分化的方向和路径。
- 识别过渡态细胞:位于不同细胞类型聚类交界处的细胞,可能就是处于状态转变过程中的中间态细胞。
TIP
报告中通常还会提供按细胞类型、样本或State拆分展示的轨迹图和细胞密度图,这些图有助于更清晰地观察特定细胞群在轨迹上的分布和富集情况。
5.2 随拟时序变化的基因
找到在轨迹上动态变化的基因是拟时序分析的另一个核心目标。
5.2.1 核心基因表达热图
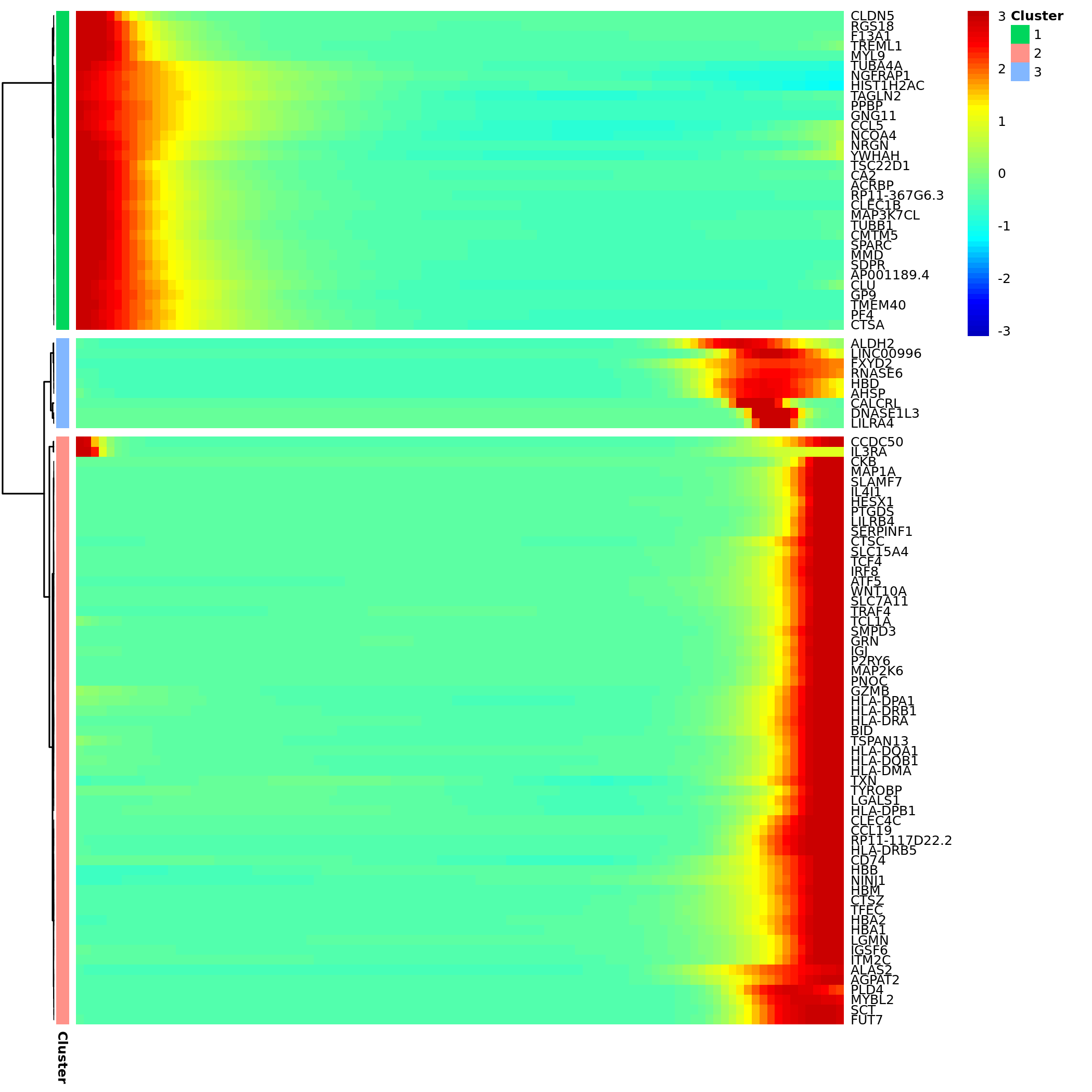
- 图表解读:热图展示了在拟时序上表达变化最显著的一组基因(通常按q-value排序)。
- 列:代表按拟时序排序的单个细胞,从左(起点)到右(终点)。
- 行:代表基因。
- 颜色:红色表示高表达,蓝色表示低表达。
- 基因聚类:左侧的聚类树将具有相似表达模式的基因聚集在一起,这些基因可能参与了共同的生物学功能。
- 分析要点:
- 识别不同的基因表达模式,例如,在发育早期高表达、晚期下调的基因(可能与干性维持相关),或是在发育晚期逐渐上调的基因(可能与终末分化功能相关)。
- 关注在分支点附近表达发生剧烈变化的基因,它们可能是细胞命运决定的“开关”基因。
5.3 分支点分析 (BEAM)
如果您的轨迹包含分支点,Monocle2会执行BEAM(Branched Expression Analysis Modeling)分析,专门寻找在细胞“做出选择”时起关键作用的基因。
5.3.1 “分支”节点基因表达热图
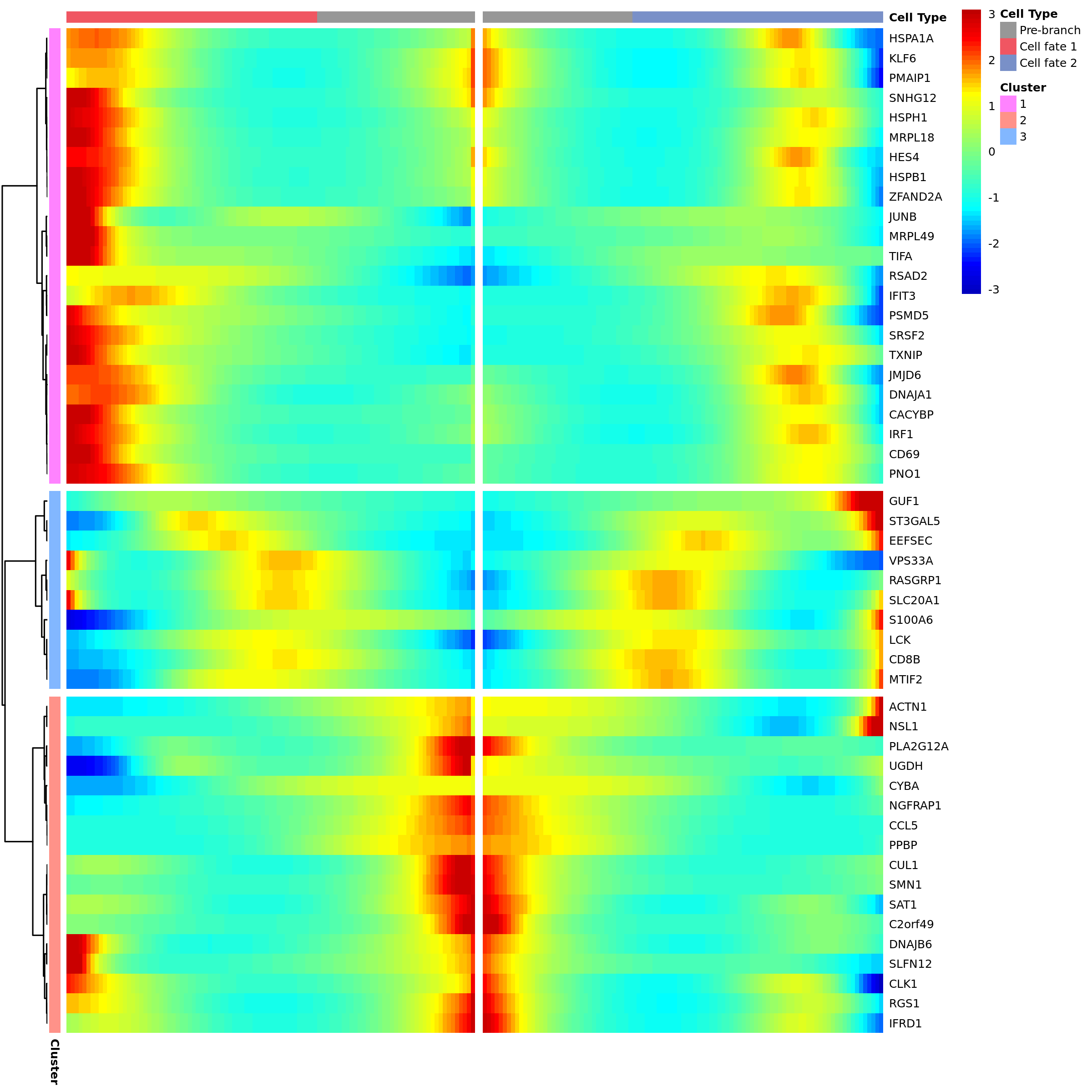
- 图表解读:该热图专门展示在通过某个特定分支点时,表达存在显著差异的基因。
- 左侧灰色区域:代表分支前的“前体”状态。
- 中间和右侧区域:分别代表细胞进入两个不同分支(Cell Fate 1 和 Cell Fate 2)后的状态。
- 分析要点:
- 寻找在一个分支中特异性高表达,而在另一个分支中低表达的基因。这些基因是定义不同细胞命运的关键因子。
- 例如,在造血干细胞分化中,一个分支可能高表达淋巴系的关键转录因子,而另一个分支则高表达髓系的关键转录因子。
5.4 功能富集分析
为了理解不同细胞状态(State)或不同基因模块的功能,报告通常会提供功能富集分析结果,如GSVA富集热图。
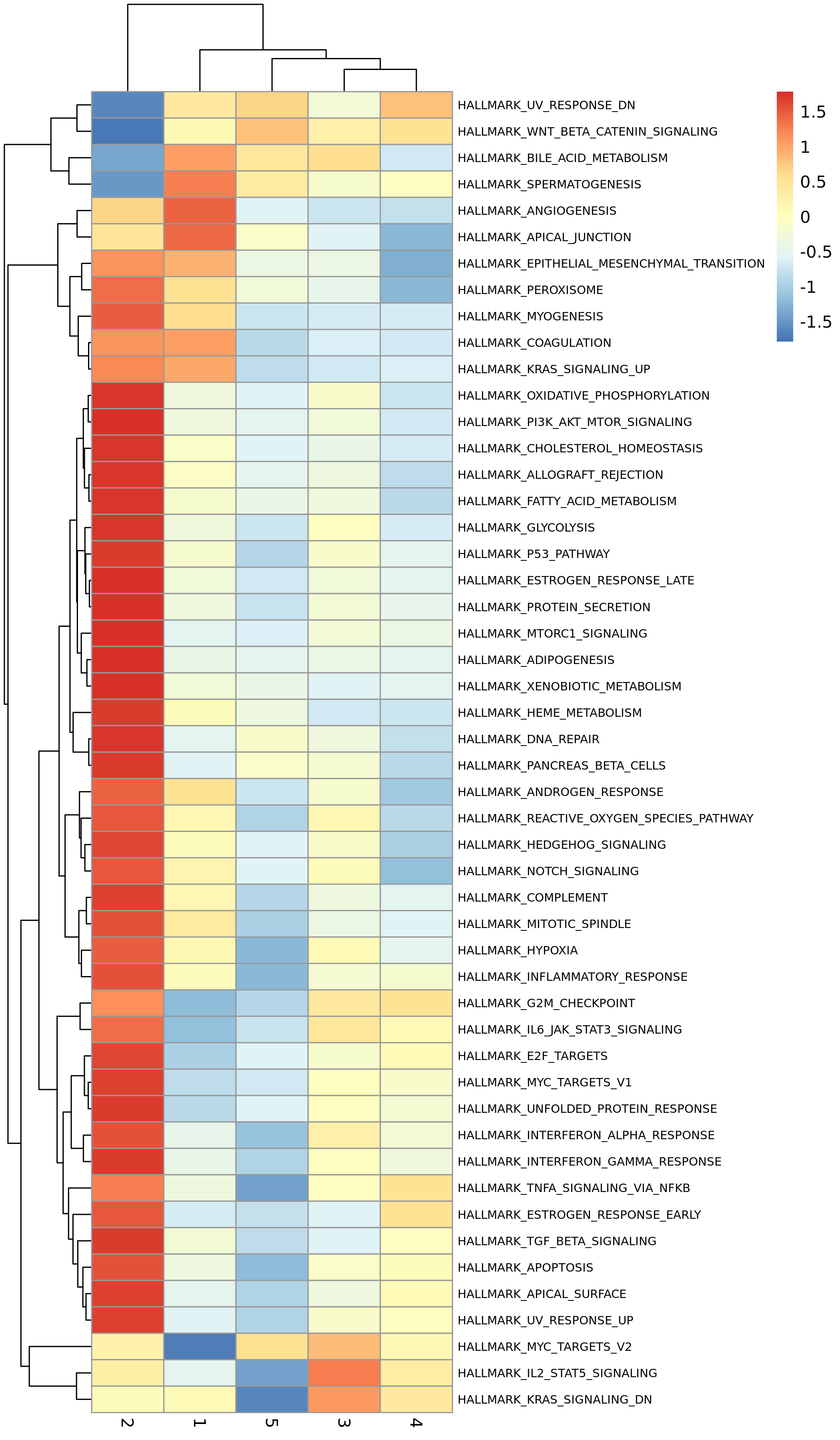
- 图表解读:热图展示了不同的生物学通路(如Hallmark通路)在不同State中的富集分数。
- 横坐标:细胞状态(State)。
- 纵坐标:生物学通路。
- 颜色:红色表示通路活性高,蓝色表示通路活性低。
- 分析要点:
- 为每个State赋予生物学功能。例如,发育早期的State可能富集“细胞周期”、“DNA修复”等通路,而发育晚期的State可能富集与特定细胞功能相关的通路,如“干扰素γ响应”(免疫细胞)或“上皮间质转换”(肿瘤细胞)。
5.5 结果文件列表
| 文件名 | 内容说明 |
|---|---|
cell_Pseudotime.csv | 核心数据:包含每个细胞的拟时序值和其所属的State。 |
ordering_genes_sig_gene_names.all.xls | 核心基因:所有随拟时序显著变化的基因列表及其统计值(p-value, q-value)。 |
branch*_pseudo_related_gene*.xls | 分支基因:BEAM分析找到的、与特定分支点相关的基因列表。 |
*trajectory*.png/pdf | 各种细胞轨迹图,包括按Pseudotime、State、celltype、Sample等不同方式着色的版本。 |
*heatmap*.png/pdf | 随拟时序变化基因或分支基因的表达热图。 |
State/GSVA/ | 各个State的GSVA富集分析结果。 |
应用案例
案例一:揭示肝癌复发中的CD8+ T细胞状态转变
- 文献:Sun Y, et al. Cell. 2021.
- 背景:研究者希望了解早期复发的肝细胞癌(HCC)中,肿瘤微环境里的CD8+ T细胞发生了怎样的变化。
- 分析策略:对来自原发灶(PT)和复发灶(RT)的CD8+ T细胞进行拟时序分析。
- 核心发现:
- 构建了CD8+ T细胞从“过渡态”到“耗竭态”的完整轨迹。
- 发现复发灶(RT)中的T细胞主要停留在轨迹的早期阶段,表现为低细胞毒性和高脂质代谢特征。
- 而原发灶(PT)中的T细胞则更多地分布在轨迹的末端,即细胞毒性或耗竭状态。
- 通过BEAM分析和基因表达动态变化分析,找到了驱动T细胞耗竭的关键转录因子和检查点分子。
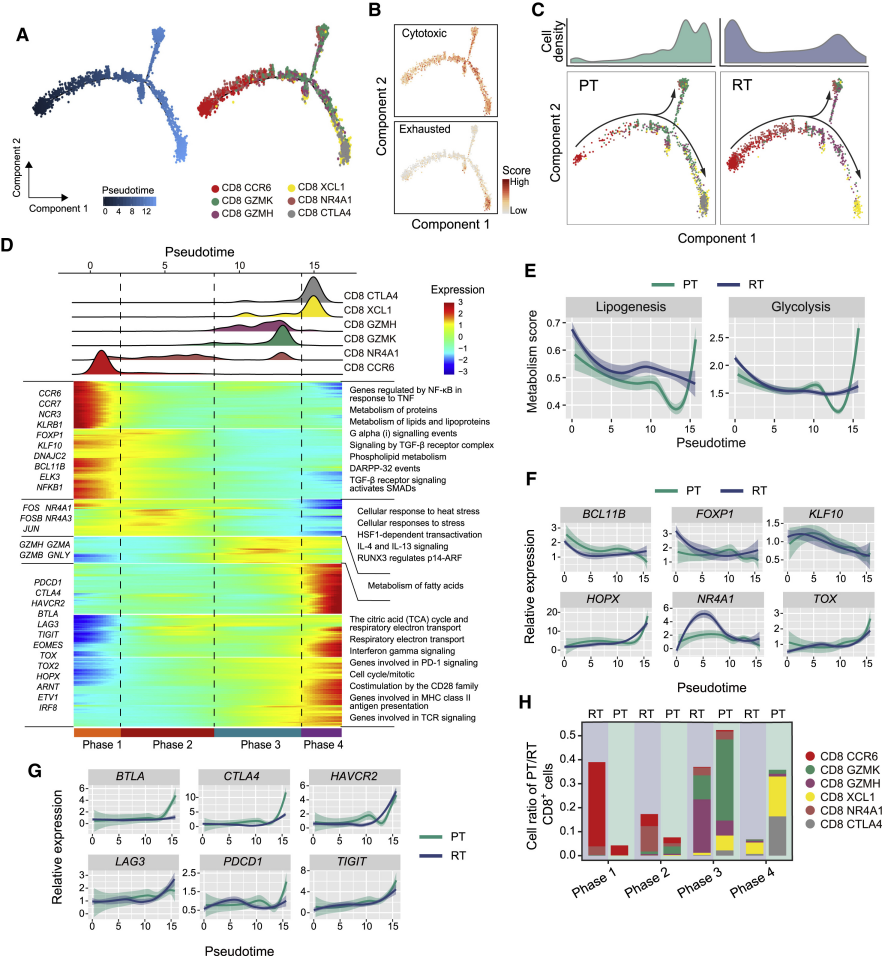 (图注:(A) PT和RT样本中CD8+ T细胞的拟时序轨迹。(C) 细胞密度图显示,RT样本的细胞(蓝色)主要在轨迹早期,而PT样本(绿色)在轨迹晚期富集。)
(图注:(A) PT和RT样本中CD8+ T细胞的拟时序轨迹。(C) 细胞密度图显示,RT样本的细胞(蓝色)主要在轨迹早期,而PT样本(绿色)在轨迹晚期富集。)
案例二:绘制人类心脏衰老的心肌细胞图谱
- 文献:Wang L, et al. Nature Cell Biology. 2020.
- 背景:研究者希望了解在心力衰竭和恢复过程中,心脏中的心肌细胞(CMs)发生了哪些变化。
- 分析策略:对来自左心室(LV)的心肌细胞进行拟时序分析,模拟其“衰老”过程。
- 核心发现:
- 构建了心肌细胞从“年轻”到“年老”的拟时序轨迹。
- 发现“年轻”的心肌细胞高表达与趋化因子和细胞因子信号相关的基因。
- 而“年老”的心肌细胞则优先表达参与脂解调节和cGMP-PKG信号通路的基因。
- 这一发现揭示了心脏衰老过程中,心肌细胞在功能和代谢上的动态转变。
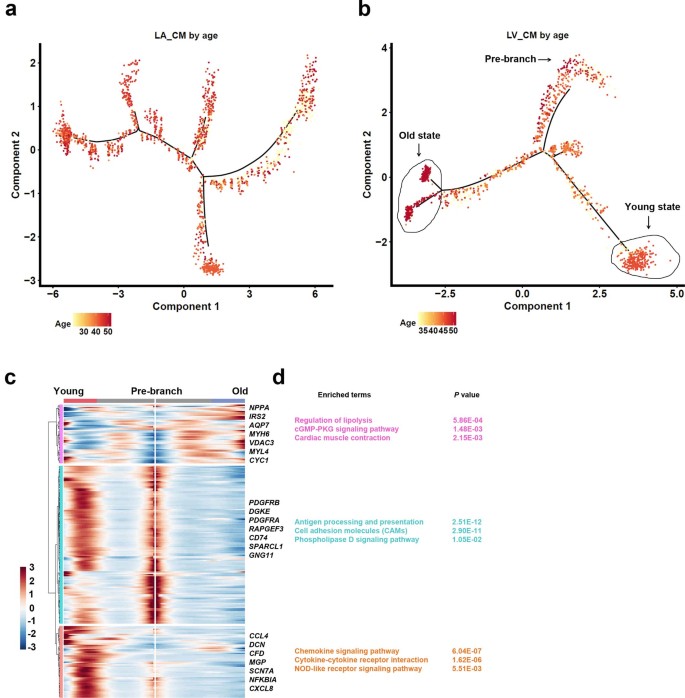 (图注:(b) LV心肌细胞的拟时序轨迹。(c) 随拟时序变化的基因表达热图,揭示了不同阶段的功能特征。)
(图注:(b) LV心肌细胞的拟时序轨迹。(c) 随拟时序变化的基因表达热图,揭示了不同阶段的功能特征。)
注意事项
1. 避免过度解读:拟时序轨迹是基于转录组数据的计算推断,不等于真实的细胞谱系。任何关键发现都需要后续的生物学实验(如谱系示踪、功能验证)来证实。
2. 起点选择至关重要:Monocle2自动推断的起点可能不正确。务必结合生物学知识,通过celltype分布图来检查和确定轨迹的生物学起点,这会直接影响整个轨迹的方向和解读。
3. 关注分支点:轨迹中的分支点是细胞命运决定的关键节点,是研究的富矿区。应重点分析BEAM的结果,找到驱动细胞走向不同命运的“开关”基因。
4. 结果不是一成不变的:拟时序分析的结果会受到上游分析(如聚类和注释)以及分析参数(如选择的细胞群、用于排序的基因)的影响。如果初步结果不理想,可以尝试调整输入的细胞群或排序基因的筛选标准,重新进行分析。
常见问题解答 (FAQ)
Q1: 图中的“State”和“Pseudotime”到底是什么意思?
A:
- State:可以理解为Monocle2根据转录组相似性自动聚类出的“细胞状态”或“阶段”。同一个State里的细胞在转录上高度相似。轨迹图上的路径连接了不同的State,展示了细胞从一个状态转变到另一个状态的过程。
- Pseudotime(伪时间):它不是真实的物理时间(小时/天),而是一个衡量细胞在分化路径上“走了多远”的抽象单位。伪时间值越小,代表细胞越接近轨迹的起点(越原始);伪时间值越大,代表细胞越接近终点(越分化)。通过分析基因表达随伪时间的变化,可以找到在分化过程中起关键作用的基因。
Q2: 如何确定哪个State是起点,哪个是终点?
A: 确定起点是结果解读的关键。请遵循以下步骤:
- 利用先验知识:查看
cell_trajectory_celltype.png图,找到您已知的、生物学上最“原始”的细胞类型(如干细胞、Naive细胞、对照组细胞),观察其主要富集的State,该State即为起点。终末分化的细胞类型所在的State则为终点。 - 利用CytoTRACE验证:CytoTRACE分值最高的细胞群,理论上对应轨迹的起点。
Q3: BEAM分析是什么?它和主轨迹图有什么关系?
A:
- BEAM (Branched Expression Analysis Modeling) 是Monocle2中一个专门用来分析分支点(Branch Point)的模块。当您的轨迹出现分支,意味着细胞在此处做出了“命运决定”。
- 作用:BEAM分析能够找出在分支点前后动态变化最显著的基因。这些基因通常是调控细胞走向不同分化命运的关键驱动因子。
- 与主轨迹图的关系:主轨迹图宏观地展示了“从哪里来,到哪里去”的路径;而BEAM分析则聚焦于“在十字路口,是什么基因决定了走哪条路”的微观问题。它是在获得主轨迹图后,进行深入机制探索的重要一步。
Q4: 排序基因是如何选择的?它为什么如此重要?
A: 排序基因是Monocle2构建轨迹的基石,它们的正确选择是获得有生物学意义结果的最关键一步。Monocle2通过观察这些基因在细胞间的表达变化模式来推断细胞的先后顺序。如果基因选择不当,轨迹将无法反映真实的生物学过程。
平台提供了三种选择排序基因的策略:
| 参数选项 | 原理 | 如何影响结果 | 适用场景 |
|---|---|---|---|
dispersion (平台默认) | 高变基因 (Highly Variable Genes):选择在所有细胞中表达水平变化最大的基因。这是一种无监督方法,它假设表达变化最剧烈的基因最可能与细胞状态转变相关。 | - 优点:无需预先定义细胞群,能进行探索性的分析,可能发现未预期的轨迹。 - 缺点:如果数据中存在强烈的技术噪音(如批次效应)或不想关心的生物学过程(如细胞周期),这些过程相关的高变基因可能会主导轨迹构建,掩盖您真正关心的过程。 | 最常用、最通用的方法。适用于大多数探索性研究,特别是当您对细胞分化的具体路径没有非常强的先验知识时。 |
differential | 差异表达基因 (Differentially Expressed Genes):找出在不同细胞群之间表达差异最显著的基因。这是一种有监督的方法。 | - 优点:目标性强,构建的轨迹将主要反映您所选定的细胞群之间的转变过程,能有效排除不相关因素的干扰。 - 缺点:结果严重依赖于您预先分组的准确性。如果分组错误,或者组间差异不能代表一个连续的过程,轨迹可能会产生误导。 | 当您有非常明确的生物学问题和清晰的细胞分群时使用。例如,您想专门研究从“干细胞”到“心肌细胞”的分化路径,就可以选择这两个细胞群,用它们之间的差异基因来构建轨迹。 |
genelist | 自定义基因列表:用户提供一个具体的基因列表,Monocle2将只使用这些基因来构建轨迹。这是监督性最强的方法。 | - 优点:可以根据充分的先验知识,强制模型关注与特定生物学过程(如上皮-间质转化、细胞凋亡)相关的基因,进行假设驱动的分析。 - 缺点:完全依赖于所提供列表的质量。如果列表不完整或包含无关基因,会直接导致轨迹偏离真实情况,且无法发现列表之外的新现象。 | 当您有非常充分的文献或实验证据,明确知道哪些基因是驱动您所研究过程的核心基因时使用。适用于验证特定的生物学假设。 |
Q5: 我的轨迹图看起来很乱,有很多断开的小片段,怎么办?
A: 这通常意味着用于排序的基因没有很好地捕捉到细胞间的连续变化,或者细胞群本身不连续。建议:
- 检查细胞群:确认您选择用于分析的细胞群之间确实存在预期的生物学连续性。
- 优化排序基因:尝试使用更严格的参数筛选差异基因,或引入您所在领域的关键标记基因进行排序。
- 检查批次效应:如果您的数据来自不同批次,请先进行去批次处理,否则批次差异可能被错误地识别为生物学轨迹。
Q6: 我的轨迹中没有出现分支,但我预期应该有,这是为什么?
A: Monocle2能否识别出分支,取决于数据本身是否包含清晰的、走向不同命运的细胞亚群。如果没有分支,可能的原因是:
- 细胞主要沿着一个线性的路径进行状态转变。
- 不同命运的细胞数量太少,信号不足以形成一个独立的分支。
Q7: 如果不同工具结果有差异,如何选择和解读?哪些结果在文章中常见?
A: 结果差异是正常的,因为它们在回答不同的生物学问题。选择和解读的关键是明确您的核心科学问题。
如何选择与解读:
- 以生物学先验知识为准绳:任何计算结果都必须符合已知的生物学事实。例如,T细胞的分化轨迹起点应该是幼稚T细胞,而不是效应T细胞。如果某个工具的结果与此相悖,那么需要谨慎对待。
- 工具互证:使用CytoTRACE来验证Monocle的起点。如果CytoTRACE得分最高的细胞群与您在Monocle中设定的起点一致,那么结果的可信度就大大增加。
- 关注核心趋势:即使轨迹细节不同,核心的生物学趋势是否一致?例如,尽管Monocle和scVelo的图看起来不同,但它们是否都指向从A细胞群到B细胞群的转变?
文章中常见的结果组合:
- 图A:Monocle轨迹树:展示细胞分化的主要路径和分支,用颜色标注细胞类型或拟时序。这是最核心、最常见的轨迹图。
- 图B:CytoTRACE得分图:在UMAP图上用颜色梯度展示CytoTRACE得分,有力地证明轨迹起点的选择是客观、无偏的。
- 图C:关键基因沿拟时序的表达热图:展示定义不同分化路径的关键基因是如何随着拟时序动态变化的,为轨迹提供基因层面的证据。
- (可选) 图D:scVelo流式图:在UMAP图上叠加RNA速度矢量箭头,展示细胞状态转变的瞬时方向,提供更精细的动态信息。
通过这样一套组合拳,您可以构建一个逻辑严密、证据充分的细胞动态变化故事。
参考文献
- Trapnell, C., Cacchiarelli, D., Grimsby, J., Pokharel, P., Li, S., Morse, M., ... & Rinn, J. L. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudo-temporal ordering of single cells. Nature biotechnology, 32(4), 381-386.
- Qiu, X., Hill, A., Packer, J., Lin, D., Ma, Y. A., & Trapnell, C. (2017). Single-cell mRNA quantification and differential analysis with Census. Nature methods, 14(3), 309-315. (Describes DDRTree)
- Sun, Y., Wu, L., Zhong, Y., et al. (2021). Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell, 184(2), 404-421.
- Wang, L., Yu, P., Zhou, B., et al. (2020). Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nature cell biology, 24(1), 1-13.