ATAC + RNA 多组学:AtaCNV 基于 scATAC 的 CNV 检测与可视化
文档概览
AtaCNV 是一种专为单细胞 ATAC-seq(scATAC-seq)数据开发的拷贝数变异(CNV)检测工具。通过处理单细胞染色质可及性测序数据,AtaCNV 能够高分辨率地揭示肿瘤细胞等复杂组织内部的遗传异质性。适用于多组学单细胞数据中的 scATAC-seq 通道,实现对肿瘤等复杂样本中细胞拷贝数状态的自动推断与可视化。
CNV 分析的意义
什么是 CNV
**拷贝数变异(Copy Number Variation,CNV)**是指基因组中较大 DNA 片段在数量上的结构变异,主要表现为染色体区域的扩增(gain)或缺失(loss)。正常情况下,人类细胞为二倍体(每条常染色体通常有 2 个拷贝);当发生扩增时拷贝数超过 2,发生缺失时拷贝数低于 2。
CNV 是肿瘤等多种疾病发生和进展的重要驱动力。传统的 CNV 分析主要基于全基因组测序(WGS)或全外显子测序(WES),只能提供群体平均水平的信息。
单细胞 CNV 分析则在单细胞分辨率下推断每个细胞的拷贝数状态,能够揭示组织内部不同细胞的基因组差异与异质性。
单细胞多组学 CNV 分析的两个方向
单细胞多组学数据用于 CNV 分析主要有两个方向:
- 方向一:基于 scRNA-seq 数据 - 利用基因表达信息推断 CNV,主要工具为 InferCNV
- 方向二:基于 scATAC-seq 数据 - 利用读段数量推断 CNV,主要工具包括 epiAneuFinder、AtaCNV、CopyscAT
TIP
主流单细胞 ATAC-seq CNV 分析工具简介
目前,常用于 scATAC-seq 数据 CNV 检测的工具有:epiAneuFinder、CopyscAT 和 AtaCNV。本指南聚焦 AtaCNV 的详细用法。如需使用其他工具,可参考 AtaCNV 与 CopyscAT 的技术支持文档获取更多信息。
适用场景与主要目的
适合的样本类型:
- 肿瘤组织样本(强烈推荐) - 包含大量 CNV 事件,可揭示肿瘤异质性和亚克隆结构
- 癌前病变或发育异常样本 - 可检测早期基因组结构变异
不适合的样本类型:
- 正常健康组织 - 大多数细胞为二倍体,缺乏显著的 CNV 事件,CNV 分析意义有限
单细胞 CNV 分析的主要目的:
- 恶性细胞识别 - 区分恶性细胞与非恶性细胞(恶性细胞通常表现出大范围的、非随机的 CNV 模式)
- 肿瘤异质性解析 - 识别具有不同 CNV 特征的亚克隆群体
AtaCNV 分析的实现
AtaCNV 工具的原理
AtaCNV 的分析流程可归纳为以下几个主要步骤:
输入与初步筛选:以百万碱基对(1 Mbp)基因组片段的单细胞读段计数矩阵作为输入,首先根据片段的可映射性和零值数量,对细胞和基因组片段进行筛选。
数据平滑与去偏差:为降低极端噪声,AtaCNV 通过对每个细胞拟合一阶动态线性模型,对计数矩阵进行平滑处理。同时执行基于细胞的局部回归,消除 GC 含量可能引起的潜在偏差。
标准化处理:
- 若存在正常细胞:将平滑后的计数数据与正常细胞数据进行标准化比较,从而从染色质可及性等混杂因素中解卷积出拷贝数信号。
- 若缺乏正常细胞:因肿瘤单细胞数据常包含大量非肿瘤细胞,AtaCNV 先对细胞进行聚类,识别出高置信度的正常细胞群组,并以其平滑深度数据为基准做标准化。
联合分段与拷贝数推断:AtaCNV 应用多样本 BIC-seq 算法对所有单细胞进行联合分割,估算每个细胞所获片段的拷贝数比率。
CNV 群组判别与进一步推断:利用拷贝数比率数据,计算每个细胞群组的 CNV 负荷评分,并据此将高 CNV 负荷的群组归类为恶性细胞。对于肿瘤细胞,AtaCNV 进一步通过贝叶斯方法推断其离散拷贝数状态。
下图为 AtaCNV 主要分析流程示意:
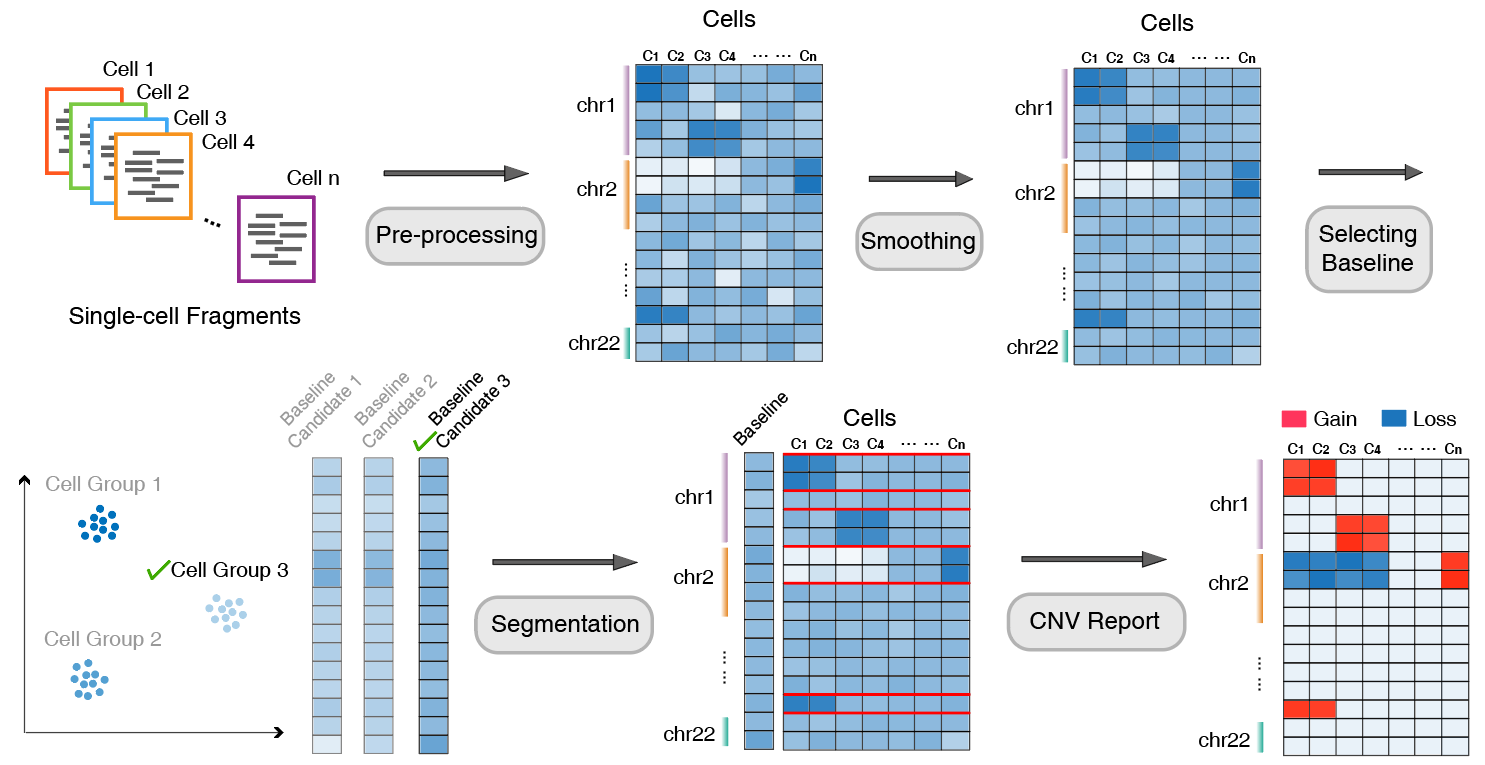
AtaCNV 分析的实现
这种方式可以精确定量每个细胞在全基因组范围内的拷贝数状态,是识别恶性细胞和解析肿瘤异质性的关键环节。实际项目落地时,建议根据数据特点选择合适的标准化模式,以获得最准确的 CNV 检测结果。
# 加载 AtaCNV 包并准备数据
library("AtaCNV")
# 从 fragment 文件和 cell barcodes 生成计数矩阵(或直接读取预处理好的数据)
fragment_file <- "your_fragment_file.tsv.gz"
cell_barcodes <- read.csv("your_cell_barcodes.tsv", header=FALSE)
count <- generate_input(
fragment_file = fragment_file,
cell_barcodes = cell_barcodes,
genome = "hg19", # 或 "hg38", "mm10"
output_dir = "./"
)
# 读取示例数据
#cell_info <- readRDS("./example/cell_info.rds")
#count_paired <- readRDS("./example/count_paired.rds") # 匹配的正常样本
# 标准化:根据可用信息选择模式
# 模式 1:匹配的正常样本(最可靠)
norm_re1 <- AtaCNV::normalize(
count,
genome = "hg19",
mode = "matched normal sample",
cell_cluster = cell_info$cluster,
count_paired = count_paired,
output_dir = "./",
output_name = "norm_re1.rds"
)
# 模式 2:已知的正常细胞
# norm_re2 <- AtaCNV::normalize(
# count,
# genome = "hg19",
# mode = "normal cells",
# normal_cells = (cell_info$cell_type == "normal"),
# output_dir = "./",
# output_name = "norm_re2.rds"
# )
# 模式 3:所有细胞(仅适用于肿瘤纯度较低的情况)
# norm_re3 <- AtaCNV::normalize(
# count,
# genome = "hg19",
# mode = "all cells",
# cell_cluster = cell_info$cluster,
# output_dir = "./",
# output_name = "norm_re3.rds"
# )
# 模式 4:自动识别(无额外信息时)
# norm_re4 <- AtaCNV::normalize(
# count,
# genome = "hg19",
# mode = "none",
# cell_cluster = cell_info$cluster,
# output_dir = "./",
# output_name = "norm_re4.rds"
# )
# 分段:使用 BIC-seq 算法推断 CNA 断点并估算拷贝数比率
seg_re <- calculate_CNV(
norm_count = norm_re1$norm_count,
baseline = norm_re1$baseline,
output_dir = "./",
output_name = "seg_re.rds"
)
# 可选:推断拷贝状态
CN_state <- estimate_cnv_state_cluster(
count = count,
genome = "hg19",
copy_ratio = norm_re1$copy_ratio,
bkp = seg_re$bkp,
label = cell_info$cell_type
)
# 热图可视化
plot_heatmap(
copy_ratio = seg_re$copy_ratio,
cell_cluster = cell_info$cluster,
output_dir = "./",
output_name = "copy_ratio.png"
)TIP
标准化模式选择
AtaCNV 提供了四种标准化模式来估计非肿瘤细胞的基线读段计数:
- 模式 1(matched normal sample):最可靠,当有匹配的非肿瘤样本时优先使用
- 模式 2(normal cells):当样本中的正常细胞已知时使用,可通过 ArchR 等工具从 scATAC-seq 数据估计标记基因表达来初步识别正常细胞
- 模式 3(all cells):仅适用于肿瘤纯度较低的情况
- 模式 4(none):当没有额外信息时使用,AtaCNV 会自动识别最可能的正常细胞,但准确性可能较低
如果没有预存在的聚类结果,可以省略 cell_cluster 参数,AtaCNV 将基于计数矩阵自动进行聚类。
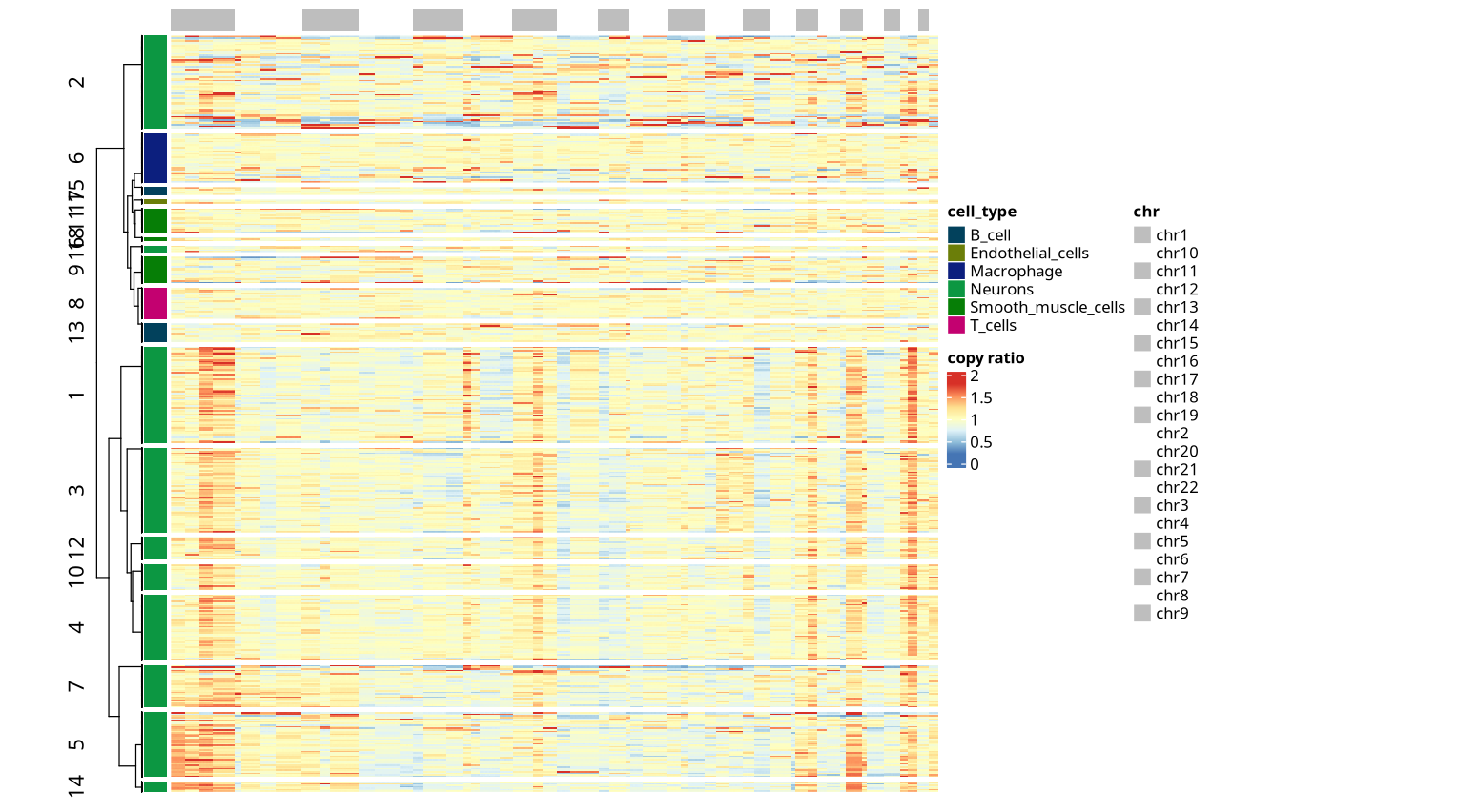
CNV 结果展示--拷贝数变异热图
拷贝数变异热图是 CNV 分析的核心可视化结果,全面展示了所有细胞在全基因组范围内的拷贝数状态:
拷贝数变异热图中,每一行代表一个细胞,每一列代表基因组上的一个连续区间(bin)。所有列按照染色体的物理位置顺序排列,从 1 号染色体依次到性染色体(若有)。每一列的颜色反映该区间在对应细胞中的拷贝数相对值:如红色表示拷贝数扩增(gain),蓝色表示拷贝数缺失(loss)。热图可见相邻染色体之间的列数可能显著不同(如有的 3 列,有的 4 列,有的只有 1 列),体现了染色体长度的细微差别。
常见问题
Q1:如何选择合适的标准化模式?
A: AtaCNV 提供了四种标准化模式,选择建议如下:
- 模式 1(matched normal sample):最可靠,当有匹配的非肿瘤样本时优先使用
- 模式 2(normal cells):当样本中的正常细胞已知时使用,可以通过 ArchR 等工具从 scATAC-seq 数据估计标记基因表达来初步识别正常细胞
- 模式 3(all cells):仅适用于肿瘤纯度较低的情况,使用所有细胞构建基线
- 模式 4(none):当没有额外信息时使用,AtaCNV 会自动识别最可能的正常细胞,但准确性可能较低
- 建议:如果可能,尽量使用模式 1 或模式 2,以获得更准确的 CNV 检测结果
Q2:如何解释拷贝状态结果中的数值?
A:在拷贝状态推断结果中:
- 0.5:表示拷贝数缺失(loss),该区域在对应细胞中的拷贝数低于正常水平
- 1:表示拷贝数中性(neutral),即正常的二倍体状态
- 1.5:表示拷贝数扩增(gain),该区域在对应细胞中的拷贝数高于正常水平
- 这些数值反映了每个细胞在每个基因组区间(bin)的拷贝数状态,可以用于识别恶性细胞和亚克隆结构
参考资料
[1] WANG X, et al. Detecting copy-number alterations from single-cell chromatin sequencing data by AtaCNV[J]. Cell Reports Methods, 2025, 5(1).