ATAC + RNA 多组学:epiAneuFinder 基于 scATAC 的 CNV 检测
文档概览
epiAneuFinder 是一种用于从单细胞 ATAC (scATAC) 数据中检测拷贝数变异 (CNV) 的算法。单细胞多组学数据中包含 scATAC-seq 信息,可以利用 epiAneuFinder 对多组学中的 scATAC-seq 做 CNV 分析,揭示肿瘤细胞异质性。
CNV 分析的意义
什么是 CNV
拷贝数变异 (Copy Number Variation, CNV) 是指基因组中较大 DNA 片段在数量上的结构变异,主要表现为染色体区域的扩增 (gain) 或缺失 (loss)。正常情况下,人类细胞为二倍体 (每条常染色体通常有 2 个拷贝);当发生扩增时拷贝数超过 2,发生缺失时拷贝数低于 2。
CNV 是肿瘤等多种疾病发生和进展的重要驱动力。传统的 CNV 分析主要基于全基因组测序 (WGS) 或全外显子测序 (WES),只能提供群体平均水平的信息。单细胞 CNV 分析则在单细胞分辨率下推断每个细胞的拷贝数状态,能够揭示组织内部不同细胞的基因组差异与异质性。
单细胞多组学 CNV 分析的两个方向
单细胞多组学数据用于 CNV 分析主要有两个方向:
- 方向一:基于 scRNA-seq 数据 —— 利用基因表达信息推断 CNV,主要工具为 infercnv
- 方向二:基于 scATAC-seq 数据 —— 利用读段数量推断 CNV,主要工具包括 epiAneuFinder、AtaCNV、CopyscAT
TIP
主流单细胞 ATAC-seq CNV 分析工具简介
目前,常用于 scATAC-seq 数据 CNV 检测的工具有:epiAneuFinder、CopyscAT 和 AtaCNV。本指南聚焦 epiAneuFinder 的详细用法。如需使用其他工具,可参考 AtaCNV 与 CopyscAT 的官方文档获取更多信息。
适用场景与主要目的
适合的样本类型:
- 肿瘤组织样本 (强烈推荐) —— 包含大量 CNV 事件,可揭示肿瘤异质性和亚克隆结构
- 癌前病变或发育异常样本 —— 可检测早期基因组结构变异
不适合的样本类型:
- 正常健康组织 —— 大多数细胞为二倍体,缺乏显著的 CNV 事件,CNV 分析意义有限
单细胞 CNV 分析的主要目的:
- 恶性细胞识别 —— 区分恶性细胞与非恶性细胞 (恶性细胞通常表现出大范围的、非随机的 CNV 模式)
- 肿瘤异质性解析 —— 识别具有不同 CNV 特征的亚克隆群体
- 克隆进化追踪 —— 通过 CNV 模式相似性推断肿瘤的克隆进化关系
epiAneuFinder 分析的实现
epiAneuFinder工具的原理
epiAneuFinder 利用 scATAC-seq 数据中比对到基因组区域的读段数量作为该区域DNA拷贝数的代理指标。为克服单细胞测序固有的覆盖稀疏性,该系统采用以下策略:
- 数据过滤:过滤低覆盖细胞,确保后续分析基于高质量的单细胞数据
- 基因组窗口划分:将基因组划分为等长窗口 (默认窗口大小为 100,000 碱基对),并量化每个窗口的比对读段数量
- 黑名单区域移除:移除 ENCODE 黑名单区域 —— 包含端粒末端、重复区域等具有系统性可比对偏差的基因组位点,以避免干扰拷贝数推断
- 低覆盖窗口过滤:针对每个数据集,epiAneuFinder 还会剔除所有细胞中零计数占比过大的窗口,以排除特定数据集中普遍低可比的基因组区域
epiAneuFinder 利用单细胞 ATAC-seq 数据整体检测流程包括以下关键步骤:
- 原始数据输入:输入经过质量控制的 scATAC-seq fragment 文件或已处理的计数矩阵
- 窗口计数矩阵生成:将基因组划分为等长窗口,计算每个细胞在每个窗口的读段计数
- 数据质量控制:过滤低质量细胞和低覆盖窗口,移除黑名单区域
- CNV推断与可视化:通过统计模型分析每个窗口的读取计数,推断出每个细胞的拷贝数变异情况,并生成可视化结果
通过使用不同的癌症scATAC-seq数据集,epiAneuFinder能够根据单细胞的CNA谱识别细胞群体中的肿瘤内克隆异质性。研究证明这些CNA谱与相同样本的单细胞全基因组测序数据推断出的结果一致。epiAneuFinder允许从scATAC-seq数据中推断单细胞CNA信息,无需额外实验,从而解锁了否则无法探索的基因组变异层面。
下图为 epiAneuFinder 主要分析流程示意:
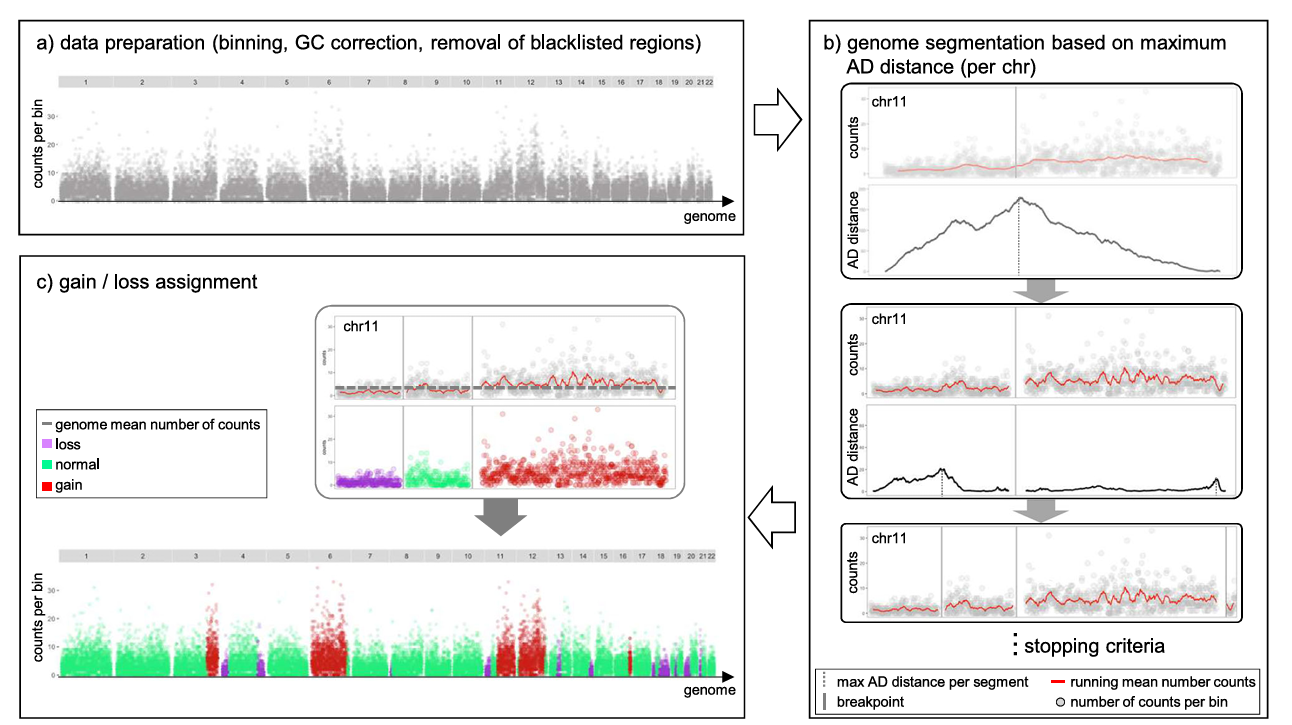
epiAneuFinder分析的实现
epiAneuFinder 可以通过以下步骤实现CNV分析。这种方式可以精确定量每个细胞在全基因组范围内的拷贝数状态,是识别恶性细胞和解析肿瘤异质性的关键环节。实际项目落地时,建议根据数据特点调整窗口大小等参数,以获得最准确的CNV检测结果。
#!/usr/bin/env Rscript
args = commandArgs(trailingOnly=TRUE)
print("Running script for epiAneufinder")
library(epiAneufinder)
# 如果使用的基因组版本不是BSgenome.Hsapiens.UCSC.hg38,请解注释下行并填入对应R包
# library(BSgenome.XXX.YYY)
# 输入数据路径,可以是fragment tsv文件,也可以是包含若干BAM文件的目录
input <- "请在此处填写输入数据的目录或fragment tsv文件名"
# 输出目录,不存在时会自动创建
outdir <- "epiAneufinder_results"
# 需在R中已安装对应的BSgenome基因组信息包,本例使用UCSC hg38基因组
genome <- "BSgenome.Hsapiens.UCSC.hg38"
# 需要排除的染色体名称,需与基因组命名一致
exclude <- c('chrX','chrY','chrM')
# 黑名单bed文件路径,需要用户根据所用基因组下载
blacklist <- "请在此处填写blacklist.bed的路径文件名"
# 基因组窗口大小,默认 1e5 (100kb)
windowSize <- 1e5
# 是否复用上一次的分析结果,默认 TRUE (部分参数变化后可能报错,酌情设置)
reuse.existing=TRUE
# 上分位数和下分位数阈值
uq=0.9
lq=0.1
# 核型图title
title_karyo="Karyoplot"
# 用于分析的CPU核心数
ncores=4
# 单细胞片段最小数量 (仅 Fragment 文件有效)
minFrags = 20000
# 如果某个 bin 在高于设定比例的细胞中都是 0 计数则过滤。0 表示关闭过滤
threshold_blacklist_bins=0.85
# 用于 CNV 推断的断点数量,1 表示全部断点。大于 1 可加速分析但降低分辨率
minsize=1
# 每条染色体的分段数 (2 的 k 次方)
k=4
# 构成CNV的最小连续bin数量
minsizeCNV=0
dir.create(outdir, showWarnings=FALSE)
epiAneufinder::epiAneufinder(
input = input,
outdir = outdir,
blacklist = blacklist,
windowSize = windowSize,
genome = genome,
exclude = exclude,
reuse.existing = reuse.existing,
uq = uq, lq = lq,
title_karyo = title_karyo,
ncores = ncores,
minFrags = minFrags,
minsize = minsize,
k = k,
threshold_blacklist_bins = threshold_blacklist_bins,
minsizeCNV = minsizeCNV
)TIP
参数调整建议
epiAneuFinder 提供了多个可调整的参数以适应不同的数据特点:
- 窗口大小 (window_size):默认 100kb,对于测序深度较高的数据可以适当减小 (如 50kb),对于测序深度较低的数据可以适当增大 (如 200kb)
- 黑名单区域:强烈建议使用 ENCODE 黑名单区域文件,以排除具有系统性比对偏差的基因组位点
- 过滤阈值:
min_cells和min_windows参数需要根据数据特点调整,过滤过严格可能丢失有用信息,过滤过宽松可能引入噪声 - CNV 推断方法:HMM (隐马尔可夫模型) 方法通常适用于大多数情况,changepoint 方法在某些情况下可能提供更好的结果
建议先用默认参数运行,然后根据结果质量进行适当调整。
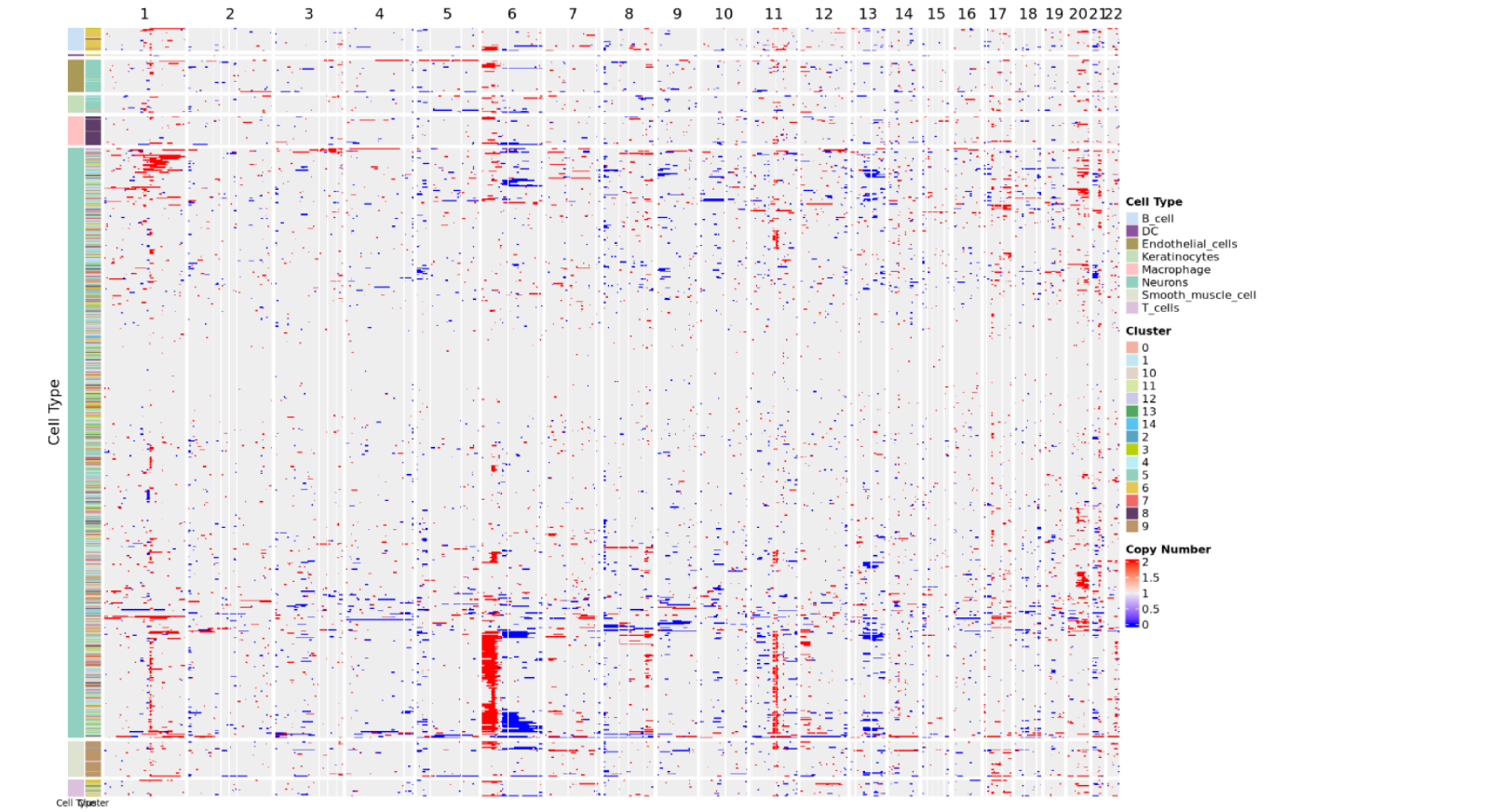
CNV 结果展示--拷贝数变异热图
拷贝数变异热图是 CNV 分析的核心可视化结果,全面展示了所有细胞在全基因组范围内的拷贝数状态:
行与列的含义:
- 每一行代表一个细胞
- 每一列代表一个染色体臂,热图的颜色表示 CNV 分数
- 列的顺序对应基因组从 1 号染色体到性染色体 (若保留) 的线性排列
颜色映射:
- copy number 值在 2 附近,表示正常的二倍体状态
- copy number 在 1~2 之间,表示染色质存在缺失,越靠近 1,表示缺失越严重
- copy number 在 2~3 之间,表示染色质存在扩增,越靠近 3 表示扩增越严重
- 颜色越红表示扩增越严重,颜色越蓝表示缺失越严重
细胞注释:
- 热图左侧色带分别表示将细胞按照细胞类型和细胞聚类情况进行排列
热图通常会在行侧添加细胞类型注释或 cluster 信息,便于区分恶性与非恶性细胞群
恶性细胞通常表现出明显的 CNV 模式 (大范围的扩增或缺失),而非恶性细胞 (如 T 细胞、B 细胞) 则保持相对正常的二倍体状态
常见问题
Q1: 如何选择合适的窗口大小?
A: 窗口大小的选择需要平衡分辨率和统计功效:
- 较小窗口 (50kb):提供更高的分辨率,但需要更高的测序深度,噪声可能较大
- 默认窗口 (100kb):适用于大多数情况,在分辨率和统计功效之间取得平衡
- 较大窗口 (200kb 或更大):适用于测序深度较低的数据,但会降低分辨率
- 建议:可以先使用默认窗口大小运行,如果检测到的 CNV 事件过多或过少,再根据数据特点调整窗口大小
Q2: 如何解释拷贝数结果中的数值?
A: 在拷贝数推断结果中:
- 接近 2:表示拷贝数正常 (二倍体状态),即该区域在对应细胞中保持正常的拷贝数
- 1~2 之间:表示拷贝数缺失 (loss),该区域在对应细胞中的拷贝数低于正常水平,越接近 1 表示缺失越严重
- 2~3 之间:表示拷贝数扩增 (gain),该区域在对应细胞中的拷贝数高于正常水平,越接近 3 表示扩增越严重
- 这些数值反映了每个细胞在每个基因组窗口的拷贝数状态,可以用于识别恶性细胞和亚克隆结构
Q3: epiAneuFinder 与其他 CNV 检测工具相比有什么优势?
A: epiAneuFinder 的主要优势包括:
- 专为 scATAC-seq 设计:充分利用了 scATAC-seq 数据的特性,直接从染色质可及性数据推断 CNV
- 无需额外实验:仅使用 scATAC-seq 数据即可检测 CNV,无需进行额外的测序实验
- 验证一致性:研究证明其 CNV 检测结果与单细胞全基因组测序数据推断的结果一致
- 参数灵活:提供多个可调整的参数,可以根据数据特点进行优化
参考资料
[1] SCHEP A N, et al. epiAneufinder: Detecting copy number alterations in single-cell ATAC-seq data[J]. Nature Methods, 2021, 18(12): 1473-1481.