单细胞 scATAC-seq & scRNA-seq 双组学标准分析:差异 Peak 分析
文档概述
差异 peak 分析是识别不同细胞群体间染色质可及性差异的关键步骤,对于理解细胞类型特异性调控机制至关重要。本文档系统介绍 peak 的概念、差异 peak 的计算方法、motif 富集分析、peak 附近基因的注释以及功能富集分析,帮助研究者全面解读细胞类型或状态间的染色质开放差异及其生物学意义。
一、核心概念
(1) 什么是reads?什么是 fragment?什么是peak?
在进行scATAC-seq数据的差异peak分析之前,我们需要先弄清楚几个核心术语:reads、fragment和peak。
1)reads:测序仪直接产出的原始数据,即一段短的DNA序列。在双端测序中,一个DNA/RNA片段会从两端各产生一条read,分别称为R1和R2。
2)fragment:实验中Tn5转座酶在基因组开放区域切割产生的原始DNA分子。双端测序中一对匹配的reads(R1和R2)可以定位出一个fragment片段。
3)peak:在基因组上,fragments的起始位点(即Tn5的切割位点)会密集地富集在开放染色质区域。这些富集区域在经过统计学分析后,被识别为一个个显著的“峰”。
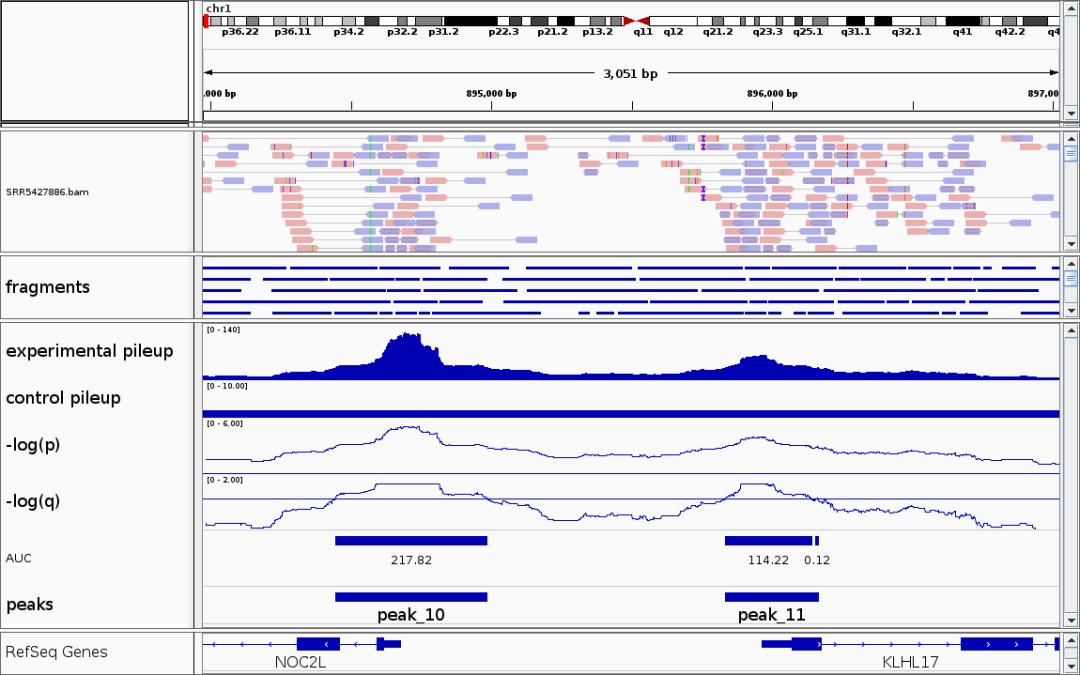
TIP
- fragmentsfragments代表了Tn5酶在基因组开放区域进行一对协同切割所产生的DNA片段,是开放染色质的直接证据。
- peaks可推断生物学功能区域。一个peak代表了一个开放染色质区域,通常与调控元件(如启动子、增强子)相关。
(2) Peaks的表示方法
与基因不同,基因名称是预先定义并统一命名的,而Peak则是在分析中(如使用 MACS 等工具 call peak 时)依据统计富集信号动态确定,因此没有统一的名称或编号。常见的标准表示方式如下,通常以“染色体-起始位点-终止位点”的格式进行标注。
chr1:9792-10697
chr1:180738-181165
chr1:181194-181660二、差异 Peak 计算:细胞群间的比较
差异 Peak 计算的核心目的是通过比较不同细胞群体(如不同细胞类型、状态、分化阶段或者疾病相关细胞)之间染色质开放区域(peak)的差异,系统性地识别那些在某一特定群体中具有更高或更低开放性的基因组区域。
(1) 差异 Peak 计算方法
以Signac为例,Signac 采用与 RNA 差异分析相同的 FindMarkers() 或 FindAllMarkers() 函数进行差异 peak 的检测,下例即为计算各细胞群相较于其它群体的差异 peaks。
# 首先设置 ATAC assay 为默认
DefaultAssay(obj) <- "ATAC"
# seurat_cluster聚类结果为例,
diff_peak_res <- FindAllMarkers(
object = obj, #假设我们已经对obj的ATAC数据做了标准化处理
group.by = "seurat_cluster", #指定细胞分群的结果, 若不指定,默认为Ident(obj)的分群结果
min.pct = 0.1, # 至少10%细胞中检测到
logfc.threshold = 0.25, # 最小log2倍数变化
only.pos = TRUE, # 只保留目标群体中更高的peak
test.use = "wilcox" # Wilcoxon秩和检验(默认)
)TIP
FindAllMarkers()适用于一次性自动比较每个细胞群体与所有其他群体的差异 peaks,非常适合多组分群(如多个细胞类型或状态)的全面差异分析。FindMarkers()适用于两组之间的直接对比(如疾病组 vs 对照组),可实现灵活的一对一差异 peaks 筛查,更便于特定分组之间的精确比较。
(2) 差异 Peak 计算的结果
下表展示的是使用 Signac 包的 FindAllMarkers() 函数对各细胞群体进行差异 peak 分析后的结果。每一行对应的是一个特定的染色质开放peak。通过这些字段,可以系统地比较各细胞群之间在不同基因组区域的开放程度,为下游功能注释和调控机制探索提供数据基础。
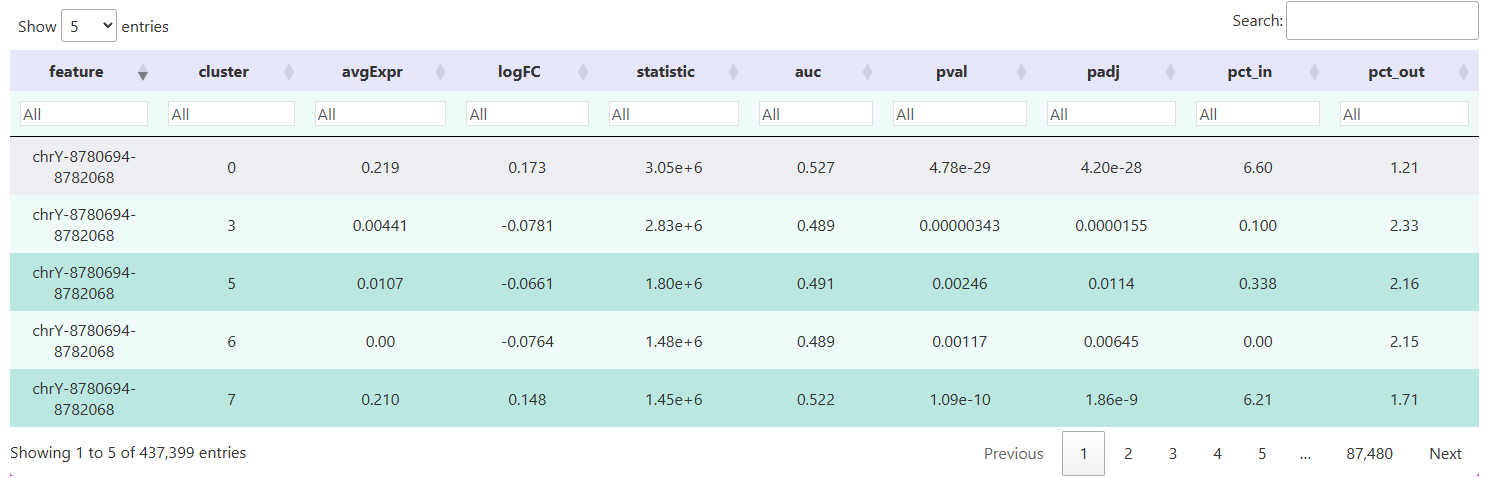
feature:差异Peak 的唯一标识符。cluster:对应细胞类别或分群标签,展示每个peak最显著开放的细胞群体。p_val:原始统计检验的P值,衡量两组细胞在该peak上的开放程度差异的显著性。p_val_adj:校正后的 P 值(通常使用 Benjamini-Hochberg 方法进行 FDR 校正)。avg_log2FC:平均对数倍数变化,反映 peak 在目标群体相对于对照群体的开放倍数(以 2 为底的对数)。pct.1:peak 在目标群体中检测到的细胞比例。pct.2:peak 在对照群体中检测到的细胞比例。
TIP
筛选标准:
- 显著性:
p_val_adj < 0.05(或更严格,如 0.01)。 - 效应量:
avg_log2FC > 0.25(或更高,如 0.5),表示在目标群体中开放程度至少是对照群体的 1.2 倍。 - 检出率:
pct.1 > pct.2,确保 peak 在目标群体中的检出率更高。
(3) 差异 Peaks 的展示
如下展示的是在cluster2细胞群中特异开放的两个peaks。差异peaks代表在一些细胞群中染色质更加开放,提示这些区域可能与细胞的功能、基因调控相关。
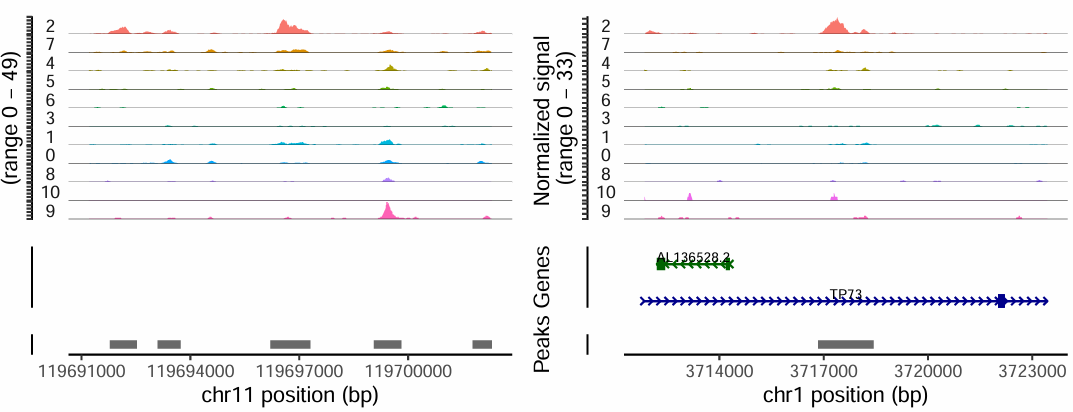
TIP
- marker基因的基因组区域也通常具有细胞群特异性的染色质开放。
- 如果某个基因是某细胞类型的marker,则其启动子或增强子附近的peak一般会在该细胞群体中特异开放。
- 我们可以通过可视化marker基因的染色质可及性,辅助细胞类型注释。
三、差异 Peaks 的 Motif 富集分析
(1) Motif 的定义
转录因子需要与DNA开放区域(peaks)结合才能发挥调控作用,每个转录因子通常有偏好的结合序列(如 SP1 偏好 GC 富集序列),这些序列模式就是 Motif,因此可以将Motif(基序)理解为转录因子结合位点的 DNA 序列模式,通常由 6-20 个碱基对组成。如下所示:

(2) Motif富集分析的目的
- Motif分析旨在通过识别差异开放peaks中富集的特定motif,推断参与调控这些peaks的关键转录因子。这有助于揭示细胞亚群间的核心调控分子,进一步解析细胞类型或状态特异性的转录调控网络及功能机制。
(3) Motif 富集分析的实现
以Signac为例, Signac使用 AddMotifs() 函数将 motif 信息添加到 Seurat 对象,并用 FindMotifs() 进行 motif 富集分析。下面是关键代码示例(以人类、hg38 为例):
library(Signac)
library(JASPAR2022)
library(TFBSTools)
# 1. 载入 motif 数据库(以人物种为例)
pfm <- getMatrixSet(
x = JASPAR2020,
opts = list(collection = "CORE", tax_group = "vertebrates", all_versions = FALSE)
)
# 2. 添加 motif 信息到 ATAC 对象(例如atac)
DefaultAssay(obj) <- "ATAC"
obj <- AddMotifs(
object = obj, # 你的Seurat/Signac对象
genome = BSgenome.Hsapiens.UCSC.hg38, # 注意加载合适的基因组包
pfm = pfm
)
# 3. 对感兴趣的peaks进行motif富集分析,使用obj中的全部peaks作为背景peaks
motif.results <- FindMotifs(
object = obj,
features = diff_peak # 例如差异分析得到的差异peaks列表
)
head(motif.results)| motif | observed | background | pct.obs | pct.bkg | fold.enrich | pvalue | motif.name | p.adjust |
|---|---|---|---|---|---|---|---|---|
| MA0497.1 | 556 | 8315 | 49.16 | 20.79 | 2.365 | 0 | MEF2C | 0 |
| MA0052.4 | 533 | 7948 | 47.13 | 19.87 | 2.372 | 0 | MEF2A | 0 |
| MA0773.1 | 398 | 4930 | 35.19 | 12.33 | 2.855 | 0 | MEF2D | 0 |
| MA0660.1 | 345 | 4002 | 30.50 | 10.01 | 3.049 | 0 | MEF2B | 0 |
| MA1151.1 | 286 | 3206 | 25.29 | 8.02 | 3.155 | 0 | RORC | 0 |
| MA0592.3 | 350 | 4669 | 30.95 | 11.67 | 2.651 | 0 | MEF2A | 0 |
motif:motif 在数据库中的编号,通常代表一个特定的转录因子的结合序列。observed:在目标 peaks(如差异 peaks)中包含该 motif 的数量。background:在背景 peaks(对照组或全部 peaks)中包含该 motif 的数量。pct.obs:目标 peaks 中带有该 motif 的百分比(= observed/目标 peaks 总数 × 100%)。pct.bkg:背景 peaks 中带有该 motif 的百分比(= background/背景 peaks 总数 × 100%)。fold.enrich:motif 在目标 peaks 中的富集倍数,= pct.obs / pct.bkg,数值越大表明 motif 在差异 peaks 中越富集。pvalue:motif 富集分析的统计显著性原始 P 值,越小意味着富集不太可能是随机出现的。motif.name:motif 对应的转录因子名称,便于直观识别调控因子。p.adjust:多重假设检验校正后的 P 值(如 FDR 校正),p.adjust < 0.05 常被认为是显著富集。
通过上述指标,可以判定哪些 motif(及其对应转录因子)在差异 peaks 中显著且强烈富集,从而揭示可能参与调控的关键 TF。
背景peaks:
- 是指在motif富集分析中用作对照的一组peaks,通常代表全体peaks或无显著差异的peaks。其作用是为感兴趣peaks(如差异peaks)中某一motif的出现频率提供一个基准。
- 通过比较差异peaks与背景peaks中某motif的富集程度,可以判断该motif是否在特定生物学状态下有显著富集。
- FindMotif()函数通过background参数可以提供一个特征向量作为背景集,或者提供一个数字来指定随机选择的特征数量作为背景集。
TIP
关于scRNA-seq+scATAC-seq双组学数据Motif分析的详细内容可查看《单细胞ATAC_RNA多组学的Motif分析》。
四、差异 Peaks 附近的基因注释
(1) Peak 与基因关联的原理(基因注释)
- Peak 本身是基因组坐标,需要通过注释将其与附近的基因关联,才能理解其生物学功能。
- 通常将 peak 与距离其最近的基因进行关联(无论距离多远)。
(2) 基因注释方法
以Signac方法为例:
# 如果对象 obj 还没有注释信息,需要先添加
# 假定已加载相关包(Signac, EnsDb, GenomicRanges等)
# 1. 添加注释信息(比如对于人物种的数据,如果已添加可跳过)
DefaultAssay(obj) <- "ATAC"
annotation <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
# 设置基因的染色体名称与obj一致
seqlevelsStyle(annotation) <- "UCSC"
genome(annotation) <- "hg38"
Annotation(obj) <- annotation
# 2. 寻找每个 差异peaks 最近的基因
closest_genes <- ClosestFeature(
object = obj,
regions = diff_peak # 或者自定义的peaks区域
)
head(closest_genes)实际输出如下:
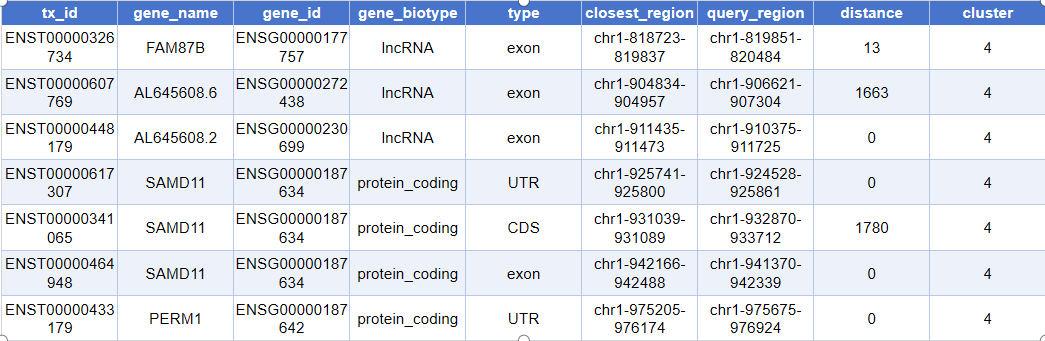
tx_id:转录本外显子/片段的唯一 ID(如 ENSE 编号)gene_name:基因的符号名称(如 TP53、GAPDH 等)gene_id:基因在数据库中的 ID(如 ENSG 开头的编号)gene_biotype:基因类型(例如 lincRNA、protein_coding、pseudogene 等)type:注释到的具体区域类型(如外显子 exon、启动子 promoter 等)closest_region:与 peak 距离最近的基因组区段(其坐标区间)query_region:被注释 peak 的基因组位置信息(坐标区间)distance:peak 到对应基因组区段最近端的距离(以碱基对 bp 计)
五、差异 Peaks 的功能富集分析
我们已通过上文完成了对 peaks 的基因注释,接下来就是对这些基因进行 GO、KEGG 等数据库的功能富集,其分析思路和流程与常规的差异基因功能富集类似。
通过对差异 peaks 注释到的基因进行功能富集,可以系统地揭示这些基因涉及的主要生物学过程和信号通路,从而为解释差异 peaks 在生物学上的潜在意义提供初步依据。
TIP
差异 peaks 的功能富集分析,本质上是对这些 peaks 所对应的邻近基因进行富集和功能注释分析。
(1) 常用功能富集数据库
GO(Gene Ontology)富集:
- 生物过程(BP):描述基因参与的生物学过程,如"细胞增殖"、"免疫应答"。
- 分子功能(MF):描述基因产物的分子功能,如"DNA 结合"、"激酶活性"。
- 细胞组分(CC):描述基因产物所在的细胞位置,如"细胞核"、"线粒体"。
KEGG 通路富集:
- KEGG(Kyoto Encyclopedia of Genes and Genomes)提供信号通路、代谢通路等注释。
- 可以帮助理解差异 peak 影响哪些关键生物学通路。
其他数据库:
- Reactome:提供详细的通路注释。
- MSigDB:包含多种基因集,如 Hallmark 基因集、细胞类型特异性基因集等。
(2) 功能富集分析示例
下图展示了针对差异 peaks 关联基因进行 GO 功能富集分析的可视化结果。纵轴显示了显著富集的 GO 条目,有助于快速识别具有代表性的功能类别及其显著性水平,从而为理解差异 peaks 可能涉及的生物学功能提供参考。z
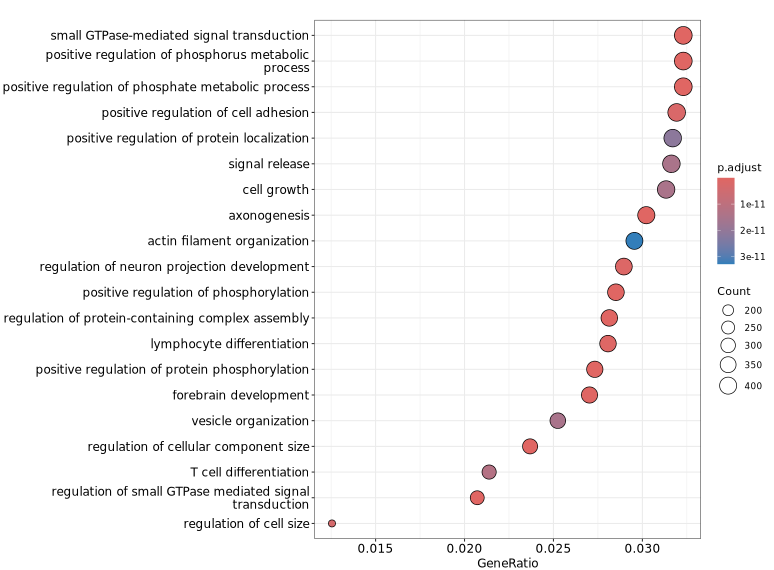
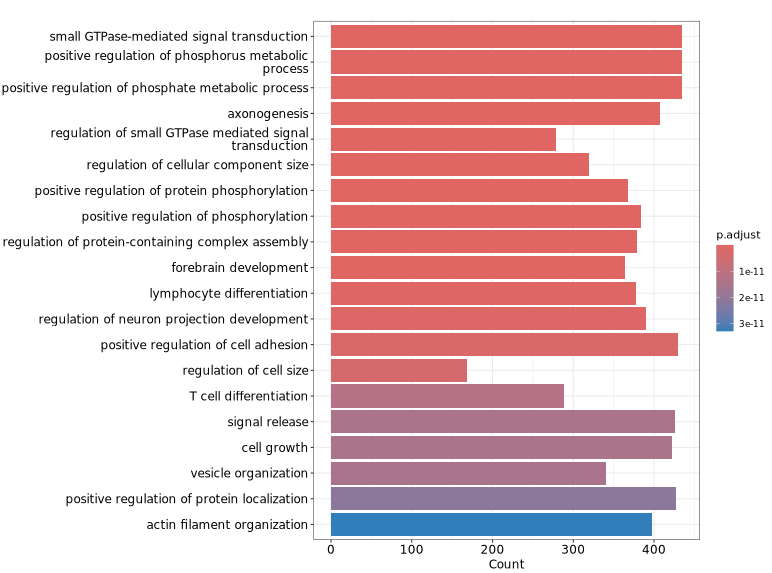
GeneRatio / BgRatio:目标基因集中包含该功能/通路的基因比例 vs 背景基因集中的比例。p.adjust:富集的显著性,p.adjust < 0.05 通常认为显著富集。Count:目标基因集中属于该功能/通路的基因数量。
TIP
Term(术语,或条目):在功能富集分析中,“term” 通常指基因集合数据库(如 GO、KEGG)中的一个功能类别,例如一个具体的生物过程(如“细胞凋亡”)、分子功能(如“DNA结合”)或通路(如“Wnt signaling pathway”)。
六、常见问题
Q1:差异 peaks 分析的目的和应用有哪些?
A:细胞群间差异 peaks 分析的主要目的是找出哪些染色质区域(peak)在不同细胞群体中开放性存在差异。这样可以帮助我们:
- 发现群体特异性的调控元件
- 揭示哪些增强子、启动子等调控区域在某一类细胞更加开放,更可能调控细胞特异性功能。
- 推测相关基因及其生物学功能
- 通过将差异 peaks 关联到附近的基因,推测这些基因的表达可能受这些调控区域影响,并进一步进行功能富集分析(如GO、KEGG)。
- 筛查潜在调控因子
- 可以结合 motif 富集等分析,预判在特定细胞群中活跃的关键转录因子。
Q2:Motif 富集分析的结果中,如何判断哪些转录因子最重要?
A:可以综合考虑以下几个方面:
前提选择:在进行 motif 富集分析时,建议优先选取差异更加显著的差异 peak(如 p 值较小或 fold-change 较高的 peaks)作为富集分析的输入集合,这样可以提高分析的生物学相关性和结果的可靠性。
指标判断:
- 富集倍数(Fold enrichment):数值越大,说明该 TF 的 motif 在 top peaks 中富集越强。
- 显著性(p.adjust):
p.adjust < 0.05(或更严格)的 TF 更可信。 - 生物学知识:结合研究背景,已知在该细胞类型或过程中重要的 TF 优先关注。
- 一致性:如果多个相关 motif 都富集同一 TF,说明该 TF 更重要。