一胞双组【ATAC+RNA】——数据分析流程、内容、项目优势
本文档系统梳理了 SeekArc 多组学单细胞数据分析的标准流程、关键质控、整合策略、高级分析及其在科学研究中的优势和应用案例。
导读
- 适用对象:单核/单细胞 Multiome(ATAC+RNA)项目用户、无生信基础入门者、技术支持与培训人员。
- 你将获得:标准流程清单、推荐质控阈值、整合与注释策略、GeneActivity 与 Peak2Gene 的原理与应用、典型论文案例、常见问题答疑。
NOTE
本文术语与流程参考 10x Genomics、Seurat、Signac 与 Human Cell Atlas 的主流实践,适配 SeekArc 项目交付与培训使用。
前提条件与数据说明
- 数据类型:单细胞/单核 Multiome(ATAC + RNA),或单组学(scRNA-seq / scATAC-seq)。
- 典型软件栈:R 生态中的 Seurat + Signac;必要时结合 Scanpy 等工具验证一致性。
- 对象与术语:文中 “ATAC” 指 scATAC-seq 可及性数据,“RNA” 指转录组表达数据;WNN 指加权最近邻(Weighted Nearest Neighbors)。
IMPORTANT
分析前建议完成样本级与批次级的质量评估与异常排查(核制备、文库、测序质量、上机深度等)。若批次差异明显,请先完成批次矫正后再进入下游分析。
基础分析
质控过滤
单细胞多组学分析的第一步是严格的质控过滤,确保后续分析的科学性和数据的可靠性。
NOTE
下述阈值为经验范围,需结合具体数据分布与项目场景灵活调整。
阈值速览(建议)
| 指标 | 建议阈值/范围 | 生物学含义 | 常见异常与处理 |
|---|---|---|---|
| nCount_RNA | 200 ~ 50,000 | RNA UMI 总数,反映转录本丰度 | 过低:空滴;过高:潜在双细胞,需结合 Doublet 检测 |
| nCount_ATAC | ≥ 1,000 | ATAC 片段事件总数,衡量开放区域覆盖 | 过低:覆盖不足;建议剔除或单独评估 |
| TSS 富集分数(Tss.enrichment) | ≥ 2 | TSS 区域开放程度与染色质完整性 | < 2 多为死亡/降解细胞,建议剔除 |
| 线粒体基因比例(mito_threshold) | ≤ 20% | 高比例提示凋亡/应激 | 高于阈值的细胞建议剔除或单独分析 |
| 核小体信号(nucleosome_signal) | ≤ 2 | 反映核小体结构完整性 | > 2 常见于降解/坏死,ATAC 质量较差,建议过滤 |
WARNING
TSS 富集 < 2 与核小体信号 > 2 往往预示着文库/细胞质量问题,继续使用会强烈影响后续降维与注释。
TIP
若整体数据质量较高,TSS 富集阈值可适当提高(如 ≥ 3),能提升后续 ATAC 层面信噪比。
常用质控指标的分布示例如下:
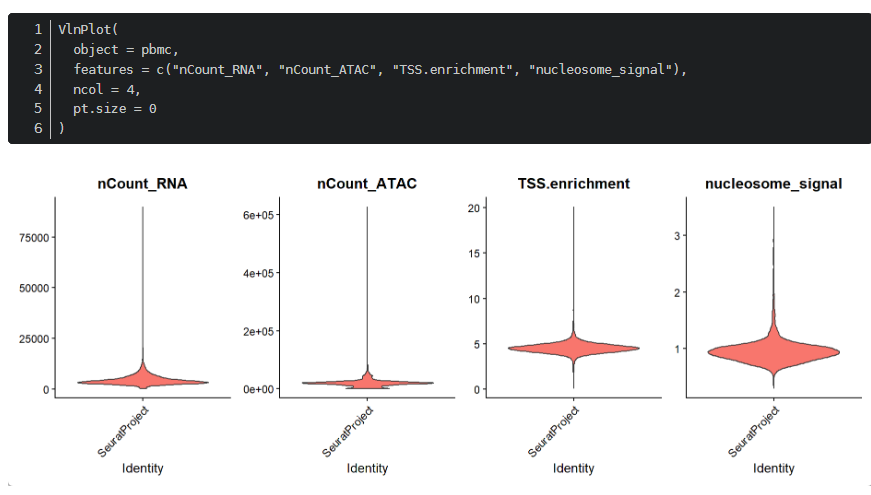 核小体信号过滤后保留的细胞和过滤掉的细胞中,fragment insert size 图的分布特征如下:
核小体信号过滤后保留的细胞和过滤掉的细胞中,fragment insert size 图的分布特征如下: 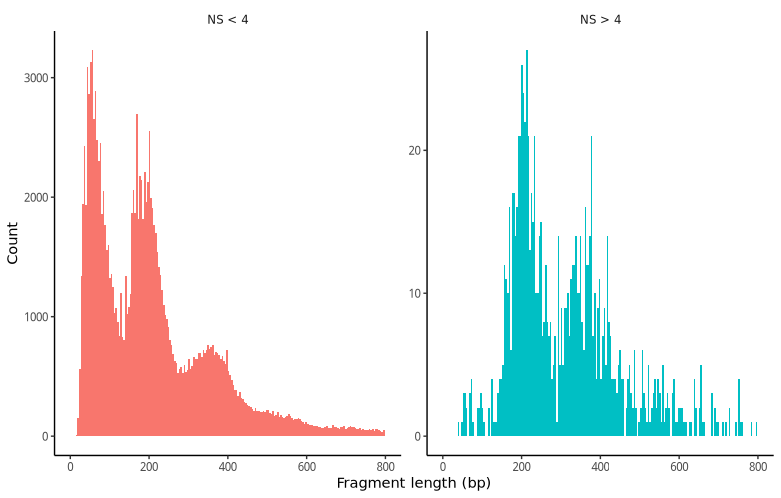
多样本整合
多样本整合旨在消除不同样本或批次间的技术差异,提升数据一致性,为后续联合分析打下基础。
scRNA-seq 和 scRNA-seq 整合
以 Seurat 的 CCA 为例,适用于不同批次或样本的 scRNA-seq 数据整合,能够有效消除批次效应,提升数据一致性。
scATAC-seq 和 scATAC-seq 整合
首先需识别各样本间的 common peaks(即在多个样本中均检测到的 ATAC 峰),随后以 Signac 的 CCA 为例进行整合,适用于 ATAC-seq 数据的降维嵌入(如 LSI)。
| 对比项 | IntegrateData (Seurat) | IntegrateEmbeddings (Signac) |
|---|---|---|
| 适用数据类型 | 主要用于 RNA 表达数据(单细胞转录组) | 主要用于 ATAC-seq 数据的降维嵌入(如 LSI) |
| 输入对象 | FindIntegrationAnchors 返回的锚点对象 | FindIntegrationAnchors 返回的锚点对象 |
| 输出内容 | 整合后的 RNA 表达矩阵(新 Assay) | 整合后的降维嵌入(新 Reduction,如 integrated_lsi) |
| 整合方式 | 校正基因表达矩阵,生成整合后的 integrated Assay | 校正降维坐标(如 LSI),生成整合后的嵌入 |
| 后续分析 | 用于 RNA 的聚类、差异分析等 | 用于 ATAC 的 UMAP 可视化、联合分析等 |
| 典型流程 | FindIntegrationAnchors → IntegrateData → 分析整合后的 RNA | FindIntegrationAnchors → IntegrateEmbeddings → 分析整合后的嵌入 |
多组学整合
- SeekArc 多样本整合:分别将 ATAC 与 ATAC、RNA 与 RNA 进行整合,方法同前述单组学整合。整合后联合 PCA 与 LSI 进行 WNN 降维。
- SeekArc 与 scRNA-seq 整合:与 scRNA-seq 之间的整合一致。
- SeekArc 与 scATAC-seq 整合:与 scATAC-seq 之间的整合一致。
IMPORTANT
ATAC 与 RNA 建议分别独立完成批次矫正与整合,再在同一对象中执行 WNN 以综合表征多组学特征。
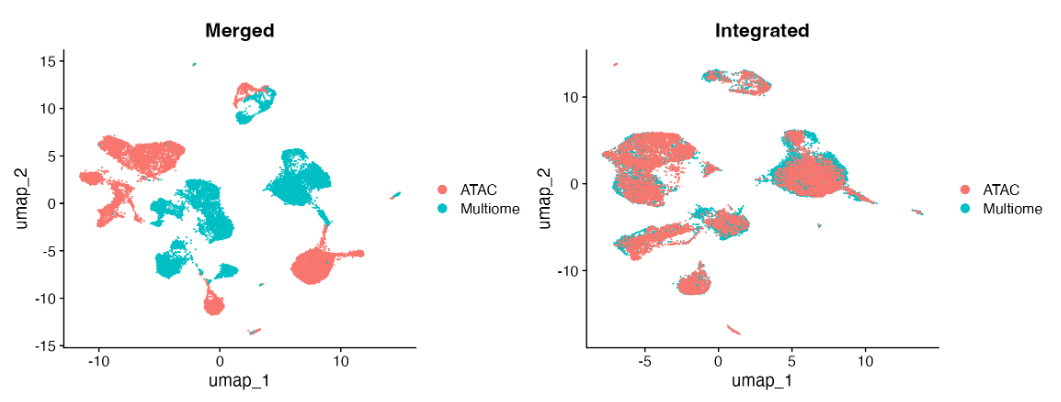
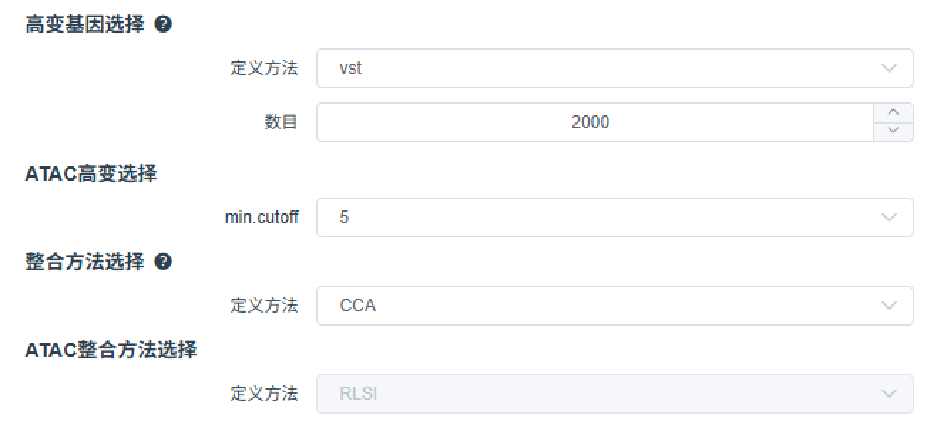
云平台多样本整合注意事项
- ATAC 高变峰选择:建议设置
min.cutoff = 5,仅将至少在 5 个细胞中均检测到开放的 peaks 纳入高变峰的计算,提高稳定性。 - 整合方法一致性:ATAC 数据的整合方法应与 RNA 数据保持一致,整合前需先计算各样本间的 common peaks,确保数据可比性。
- 批次矫正原则:ATAC 和 RNA 数据需分别独立进行批次矫正和整合。多样本整合后,数据结构仍为一个 ATAC assay 和一个或多个 RNA assay(如采用 CCA/RPCA 整合,还会生成 integrated assay)。
TIP
可在云平台一键执行常用整合流程:SeekSoul Online
降维聚类
降维聚类是多组学数据分析的核心步骤,能够揭示细胞间的异质性和潜在亚群。
- ATAC + RNA 联合聚类:采用 PCA 与 LSI 联合降维,综合两组学特征,实现对细胞异质性的全面刻画。
- RNA 聚类:基于 RNA 表达矩阵,利用 PCA 降维后进行聚类。
- ATAC 聚类:基于 ATAC 开放性数据,采用 LSI 降维后聚类。
NOTE
联合聚类(WNN)常能发现单一组学难以分辨的亚群,建议与单组学聚类结果交叉验证。
RNA 降维聚类结果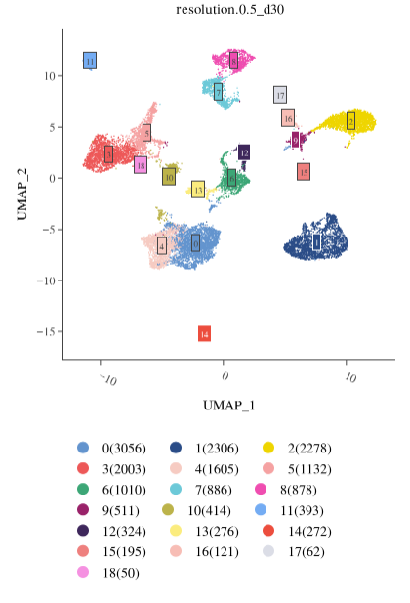 ATAC 降维聚类结果
ATAC 降维聚类结果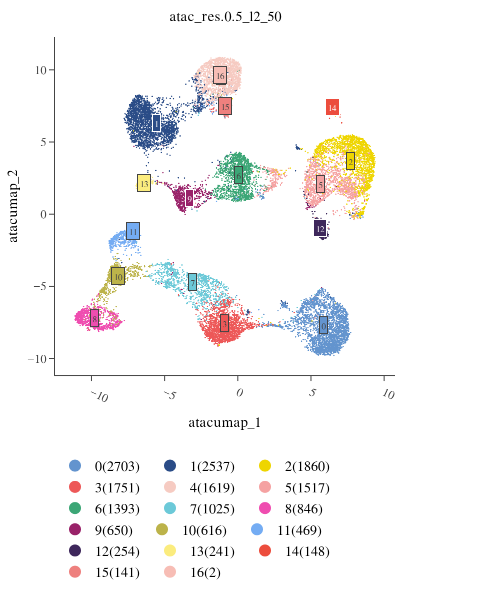 联合降维聚类结果
联合降维聚类结果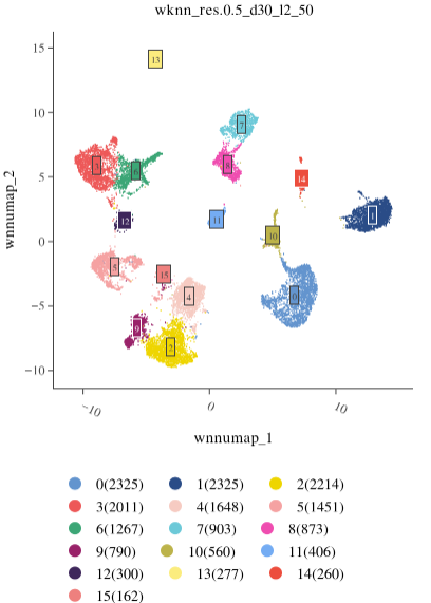
细胞注释
细胞注释是多组学分析中将聚类结果赋予生物学意义的关键环节。
- SeekArc 项目常用 WNN 聚类结合 marker 基因表达进行细胞类型判定。
- scATAC-seq 单组学注释方式包括:1)基于 marker 基因的基因活力进行注释;2)与同组织 scRNA-seq 样本映射注释(label transfer)。
IMPORTANT
注释需综合多证据:marker 表达/活力、已知参考图谱、空间/实验验证以及跨数据集一致性。
注释结果在 RNA 降维图中的分布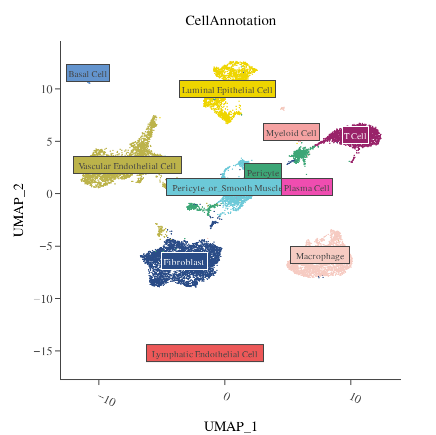 注释结果在 ATAC 降维图中的分布
注释结果在 ATAC 降维图中的分布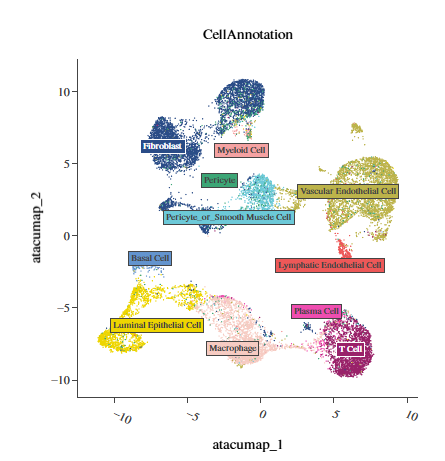 注释结果在 WNN 降维图中的分布
注释结果在 WNN 降维图中的分布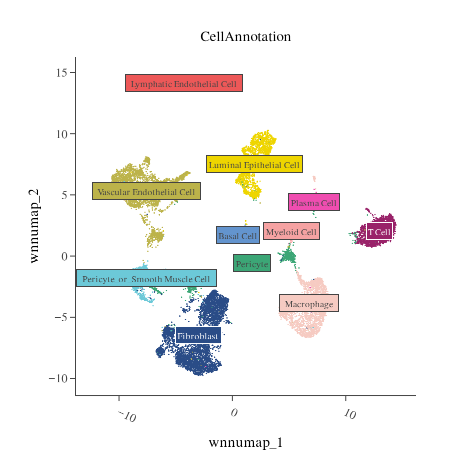
GeneActivity 高级分析
原理
GeneActivity 分析通过统计每个基因区域内的 ATAC 片段数,并结合每个细胞的总片段数,计算基因活力分数。该分数反映了基因在染色质开放层面的潜在表达能力,为后续细胞类型注释和功能分析提供依据。
GeneActivity(
object,
assay = NULL,
features = NULL,
extend.upstream = 2000,
extend.downstream = 0,
biotypes = "protein_coding",
max.width = 5e+05,
process_n = 2000,
gene.id = FALSE,
verbose = TRUE
)核心计算公式:
Gene Activity = log10((N_gene/N_total) × 10^6 + 1)
N_gene = 基因区域的片段计数
N_total = 细胞总测序片段数
基因结构图如下: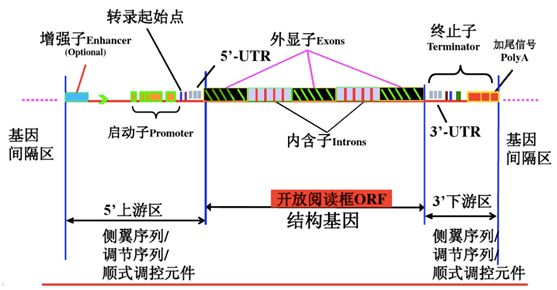
CAUTION
物种基因注释与基因坐标需与参考基因组版本严格匹配;若参考不一致会产生系统性偏差。
应用
应用一:通过 marker 基因活力对细胞进行大类注释
- 以 scRNA-seq 注释结果为参考,通过细胞标签转移(label transfer)方法,将 scRNA-seq 的细胞类型信息映射到 scATAC-seq 细胞,实现多组学间的一致性注释。
marker 基因活力情况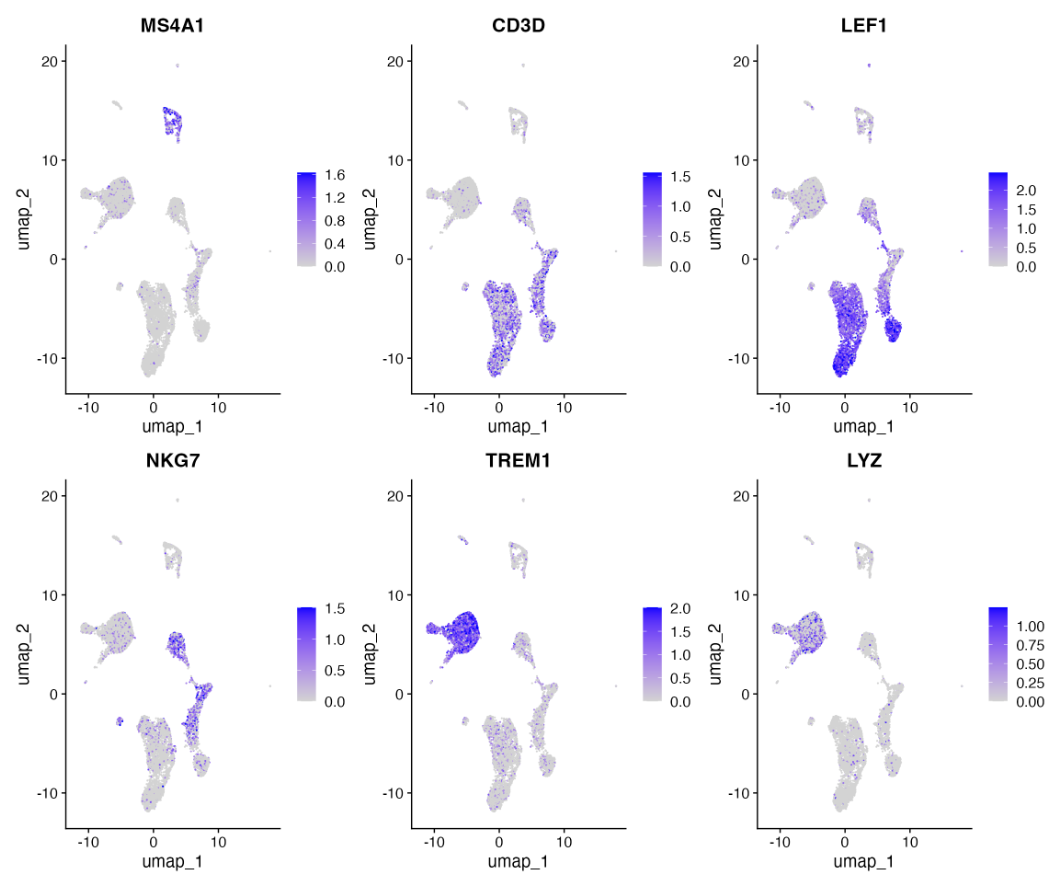 scRNA-seq 数据以 marker 基因表达情况对细胞进行注释的情况
scRNA-seq 数据以 marker 基因表达情况对细胞进行注释的情况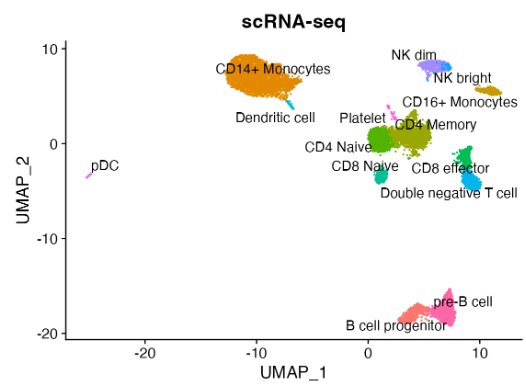 将上图中 scRNA-seq 注释结果 transfer 到 scATAC-seq 数据中
将上图中 scRNA-seq 注释结果 transfer 到 scATAC-seq 数据中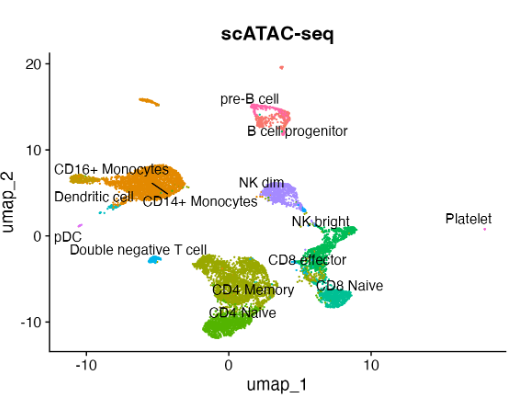
应用二:scATAC-seq 的异质性与基因活力和 scRNA-seq 的一致性
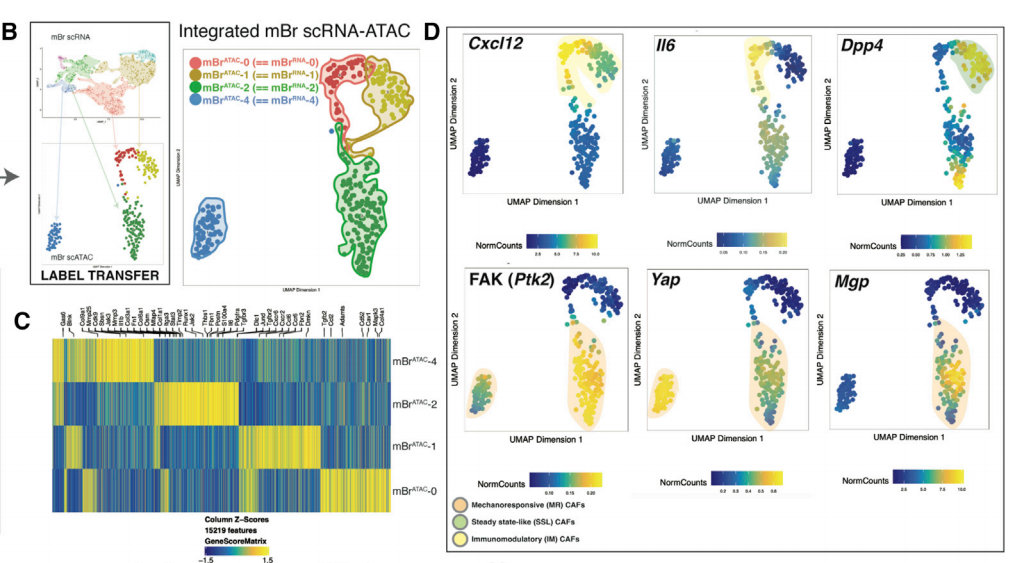
- 文章一:scATAC-seq 数据分析时,计算基因活力在 cluster 之间的差异,体现 scATAC-seq 数据具有异质性。 Multiomic analysis reveals conservation of cancer-associated fibroblast phenotypes across species and tissue of origin." Cancer cell 40.11(2022):1392-1406.e7.
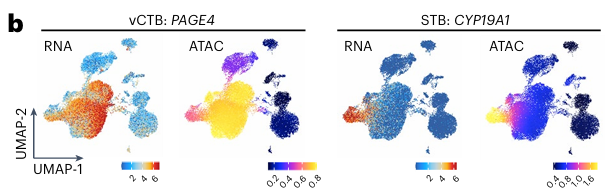
- 文章二:Single-cell multiomic 数据中,marker 基因 PAGE4 和 CYP19A1 分别在 scRNA-seq 和 scATAC-seq 数据中的表达情况和基因活力是否一致。结果一致,说明 Multiome 中的 ATAC 部分可靠。 Johain R. Ounadjela, Ke Zhang. , et al. "Spatial multiomic landscape of the human placenta at molecular resolution." nature medicine.
ATAC_Peak2Gene 高级分析
原理
通过 LinkPeaks 函数,计算 ATAC 峰与基因表达的相关性,揭示染色质可及性与基因表达的调控关系。
LinkPeaks(
object,
peak.assay,
expression.assay,
peak.slot = "counts",
expression.slot = "data",
method = "pearson",
gene.coords = NULL,
distance = 5e+05,
min.distance = NULL,
min.cells = 10,
genes.use = NULL,
n_sample = 200,
pvalue_cutoff = 0.05,
score_cutoff = 0.05,
gene.id = FALSE,
verbose = TRUE
)- 对每个位于有效距离内的峰-基因对,计算 ATAC 峰计数与基因表达量的相关性(默认 Pearson)。 如下图表示 MS4A1 基因与多个 peaks 具有相关性
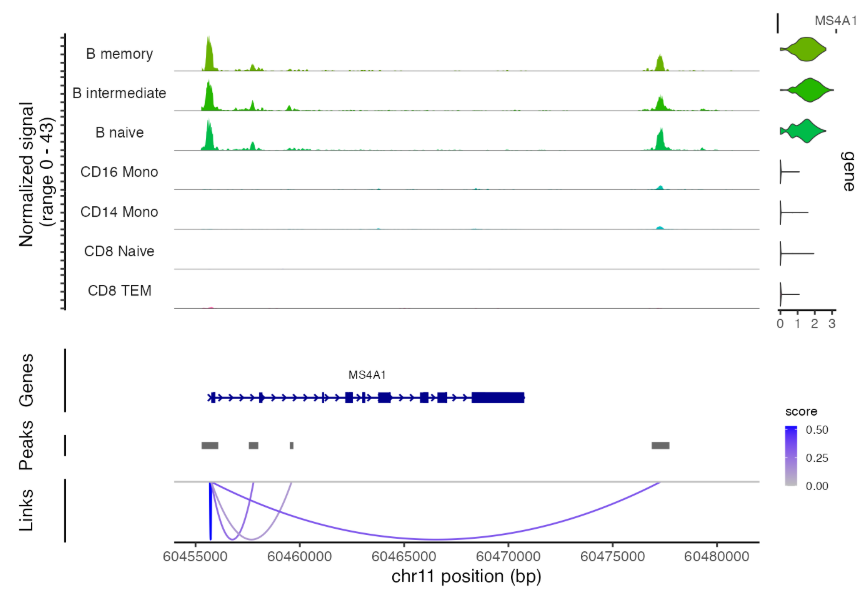
WARNING
相关性并不等价于因果调控。建议结合 motif 富集、footprinting、转录因子活性评分与外部先验进行交叉验证。
应用
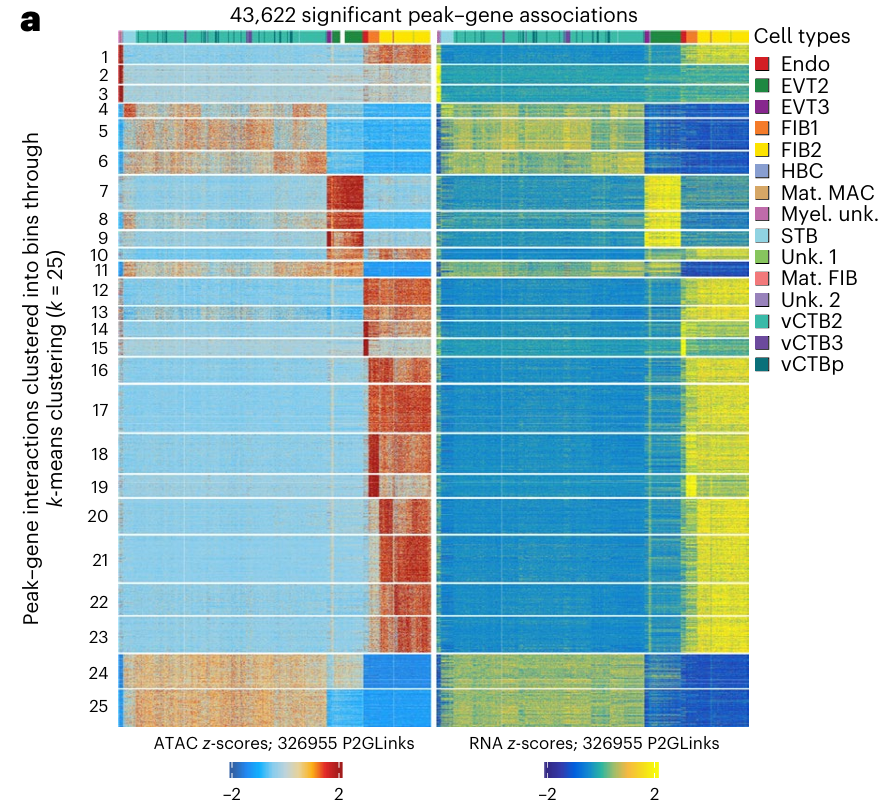
应用一:识别 DORC 与关键调控因子
- 通过 genes-peaks links 分析,找到 DORC 区域,发现细胞类型特异性 DORC 及关键调控因子(如 TBX5)。 *Johain R. Ounadjela, Ke Zhang. , et al. "Spatial multiomic landscape of the human placenta at molecular resolution." nature medicine. *
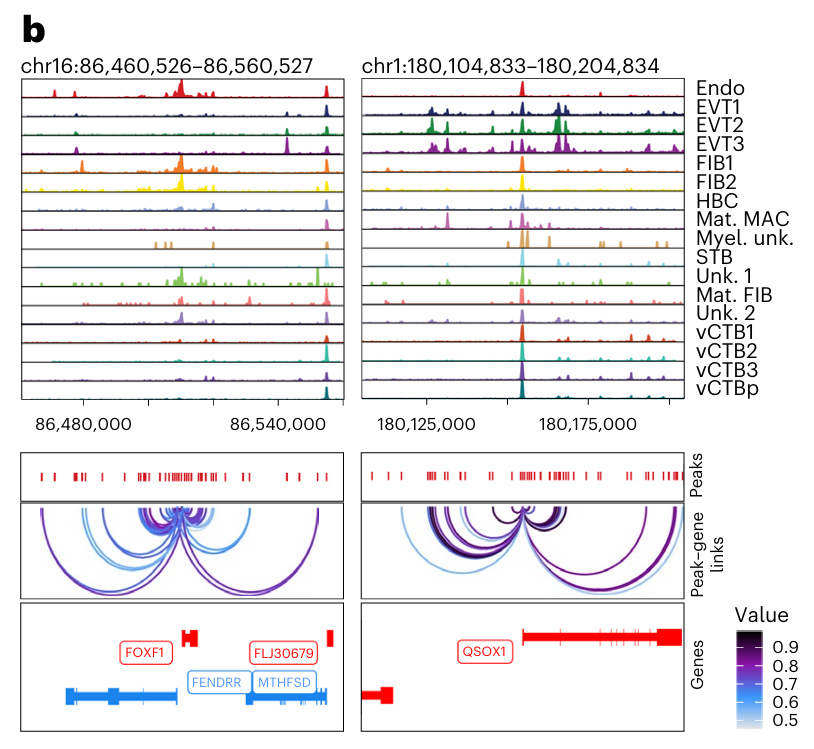
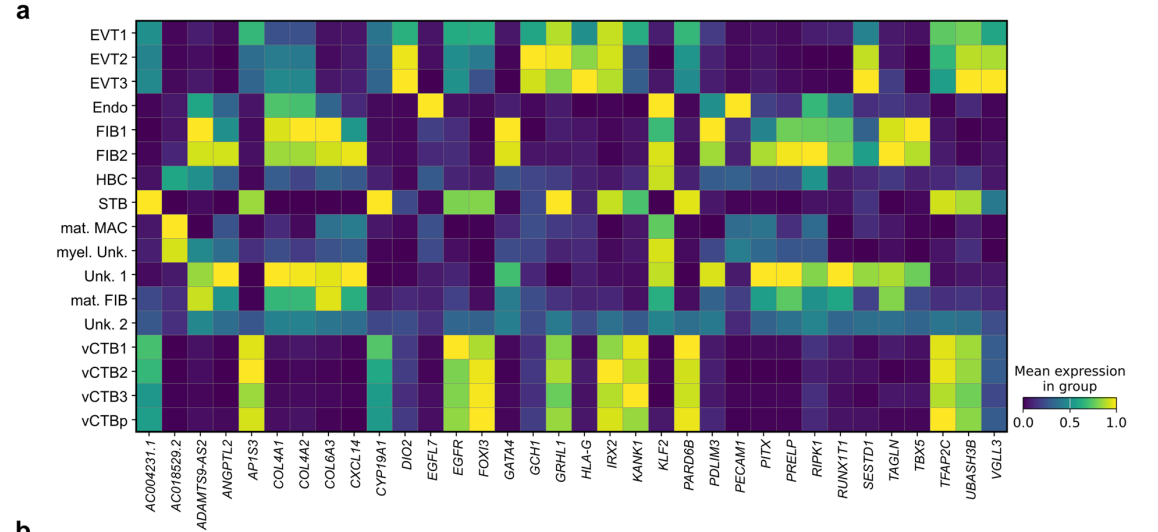
应用二:差异峰的 motif 富集与 TF 识别
- 对差异 peak 做 motif 富集分析,识别关键转录因子(如 RUNX1)及其靶基因,探究其在疾病中的作用。 Granja, Jeffrey M. , et al. "Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia." Nature Biotechnology
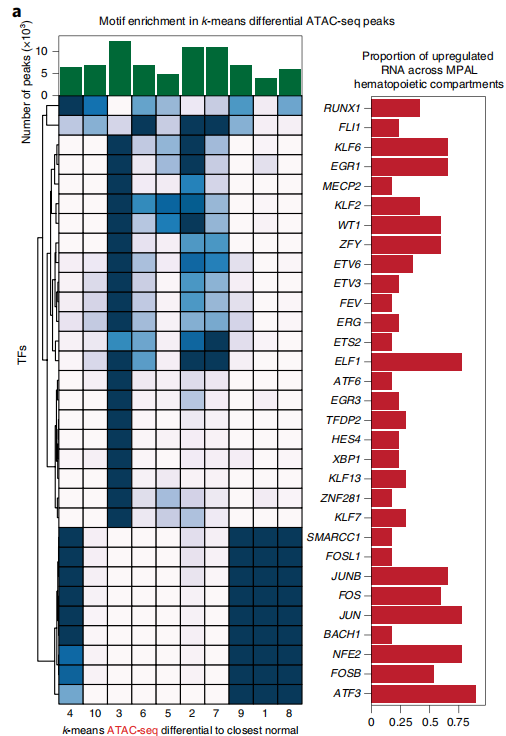
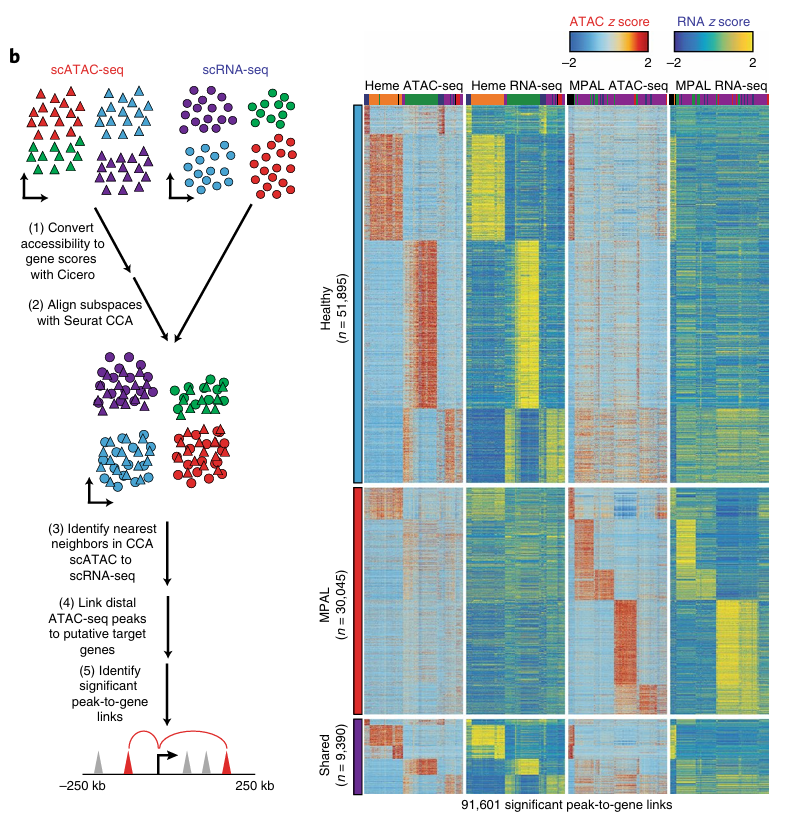
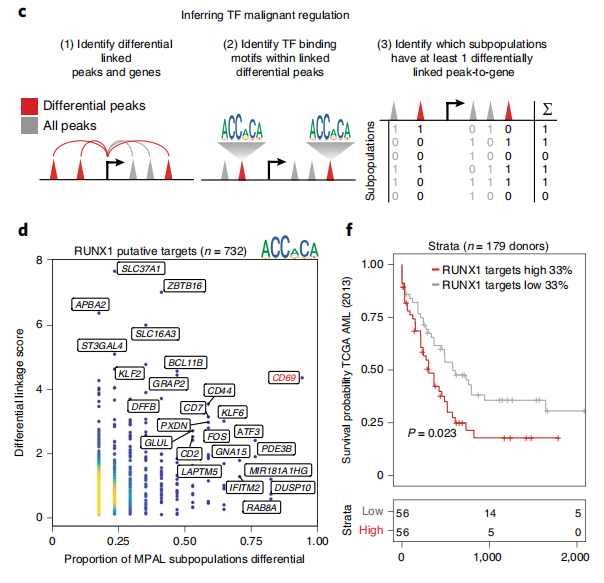
数据分析优势与案例
利于新亚类发现
文章 1
Hao, Yuhan , et al. "Integrated analysis of multimodal single-cell data - ScienceDirect." Cell 184.13(2021):3573-3587.e29.
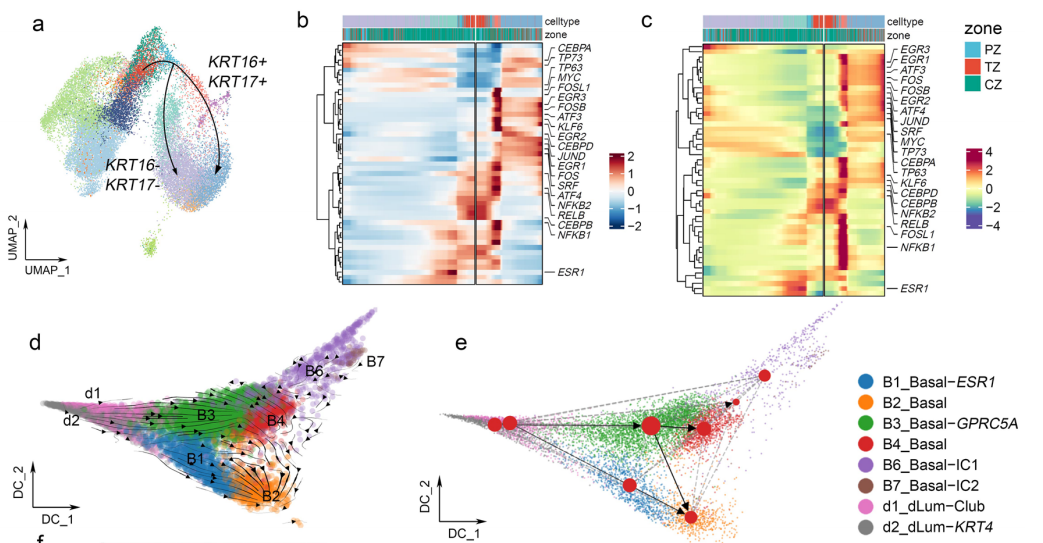
- ATAC 的异质性比 RNA 更强。在 Multiome 的 WNN 降维聚类中,发现了 Basal_4 亚类。
- Basal_4 类和 Basal_1 类转录组高度相关,因此在 RNA 降维中,Basal_4 未能独立成簇。
- Basal_4 在 ATAC 层面具有显著特异性,在 ATAC 中能独立成簇,但难以通过传统注释方法识别,只有在 WNN 联合分析中才能被发现和注释。
- 结论:WNN 能灵敏、稳健地表征单一组学数据无法识别的细胞群体。
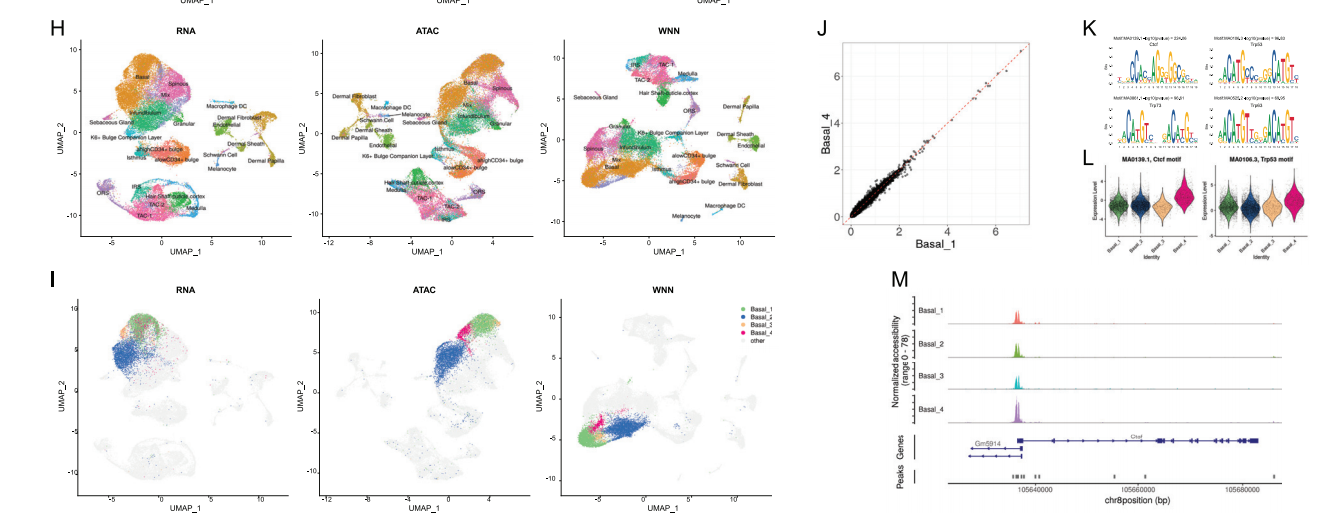
文章 2
"Spatially resolved transcriptomic analysis of the adult human prostate." Nature Genetics.
- ATAC 的异质性比 RNA 更强。分析前列腺基底细胞 Basal cell 时,RNA 层面无明显差异,但 ATAC 层面可分为 KRT16+KRT17+ 和 KRT16−KRT17− 两种亚型。
- 发现 B3、B4、B5、B6 亚类 KRT16 与 KRT17 开放性更强,将其注释为 KRT16+KRT17+ 基底细胞,其余为 KRT16−KRT17− 细胞。
- 拟时序分析显示,这两类细胞是基底细胞的两个分化方向,具有真实生物学意义。
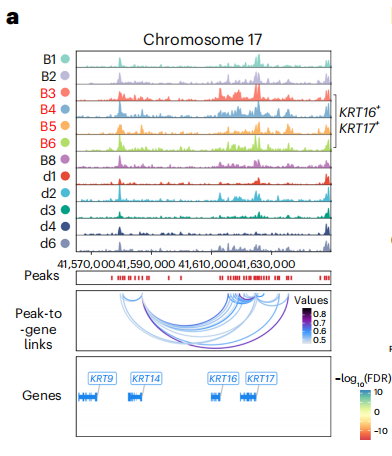
数据分析优势
文章 1
"Single-cell transcriptomic and chromatin dynamics of the human brain in PTSD." Nature.
- 利用 scATAC 与 scRNA 单组学做 gene-peak 共可及性分析,发现 PTSD 改变了基因表达的顺式调控元件。用 Multiome 数据验证,提示单组学做 gene-peak 关联存在风险,需要 Multiome 验证。 scATAC-seq+scATAC-seq 单组学分析结果
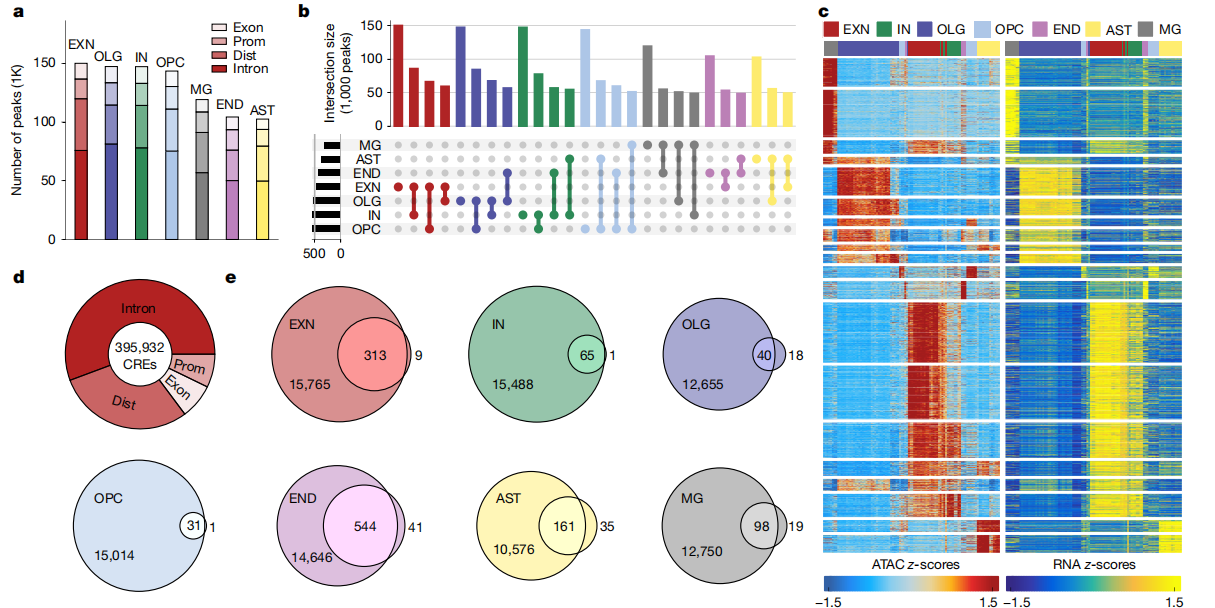 multiomic 共开放性分析
multiomic 共开放性分析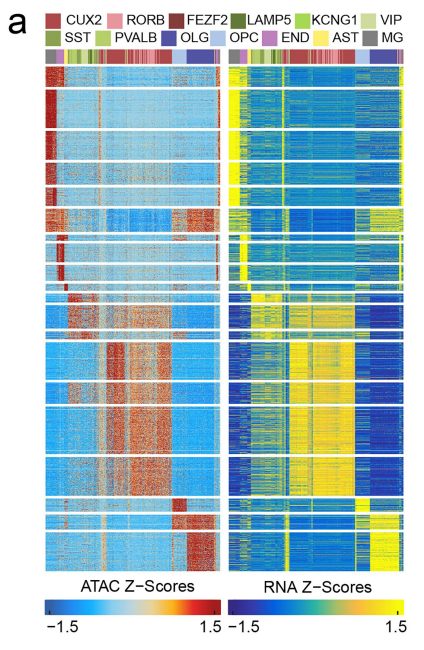
文章 2
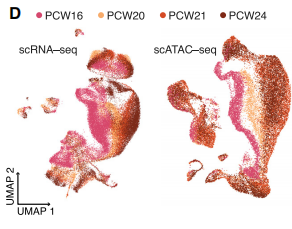
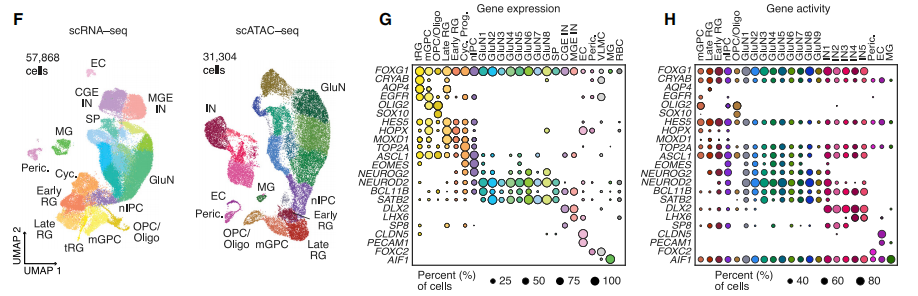
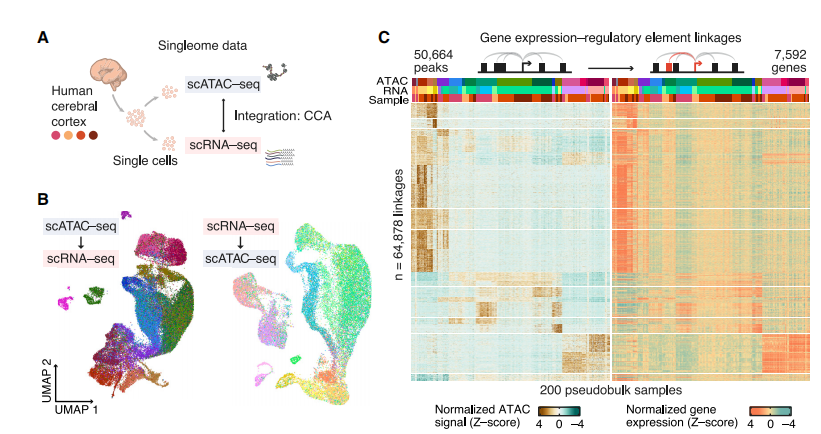
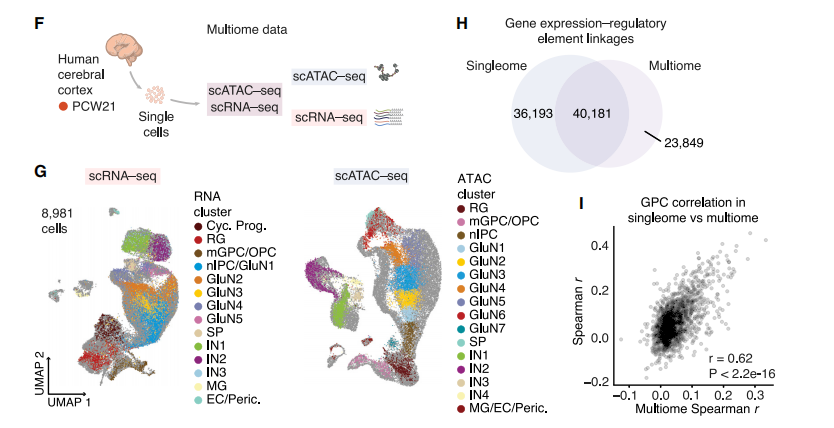
Trevino, Alexandro E. , et al. "Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution." Cell D1(2021).
- 先分别用 scRNA-seq 与 scATAC-seq 单组学,基于基因表达和基因活力完成细胞注释。 开始做了 scRNA-seq 和 scATAC-seq 单组学,分别根据基因表达和基因活力完成细胞注释
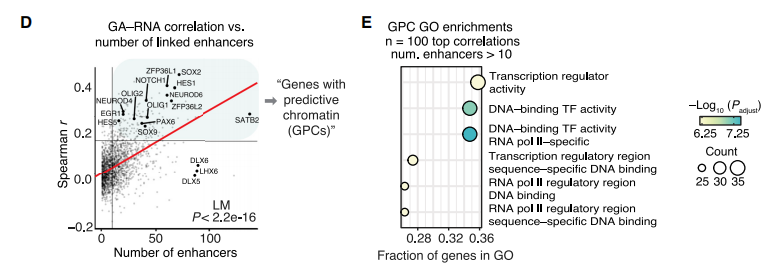
- 进一步发现 GPC 调控关系,它们可能是细胞命运决定的关键调控因子。 发现 GPC 调控关系,它们可能是细胞命运决定的关键调控因子
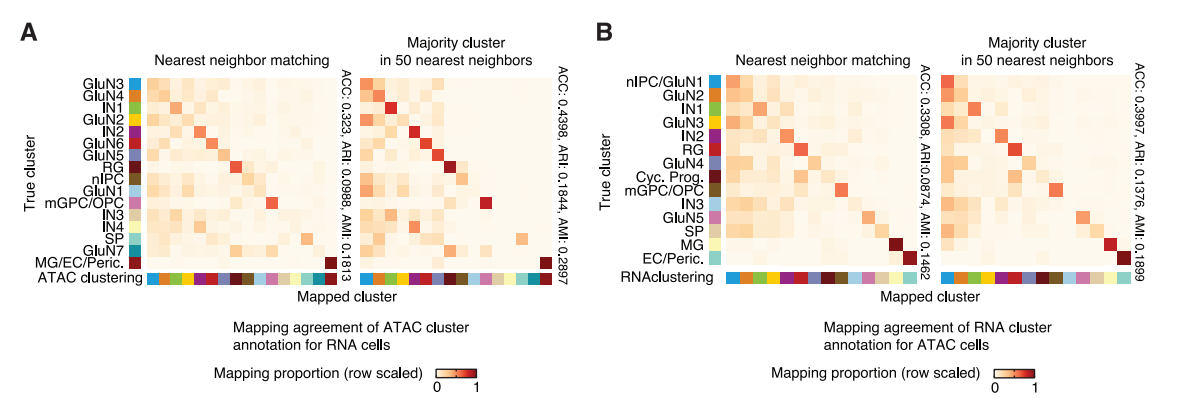
- 用一胞双组学对前面单组学中的 peaks-genes Links 进行对比,两者算出来的 Links 有较大差异,前后算出来的 GPC 具有较好的一致性。 比较一胞双组学和单组学中分析 peaks-genes Links 的 overlap 和相关性
- 将 CCA 算法用到 Multiome 内部,验证了标签映射效果受参数影响,也侧面说明 Multiome 同时测表达与开放的准确性是单组学难以达到的。 CCA 参数对细胞标签 transfer 结果的影响
文章 3
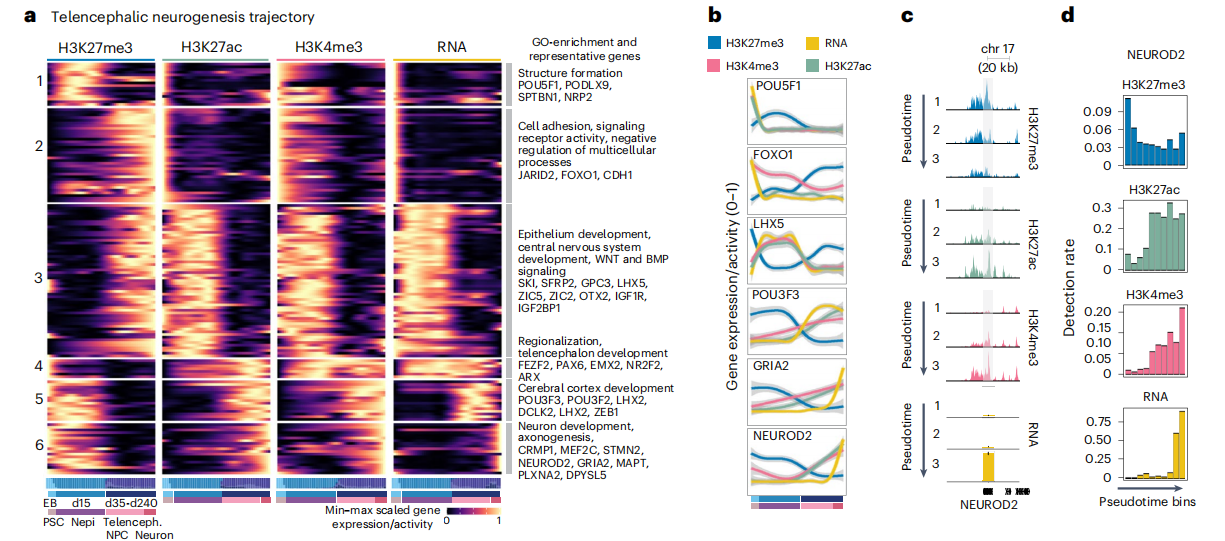
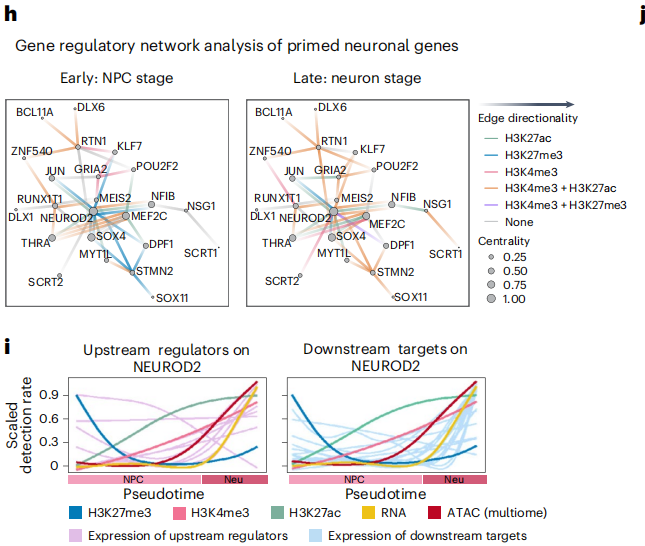
Fides, Zenk , et al. "Single-cell epigenomic reconstruction of developmental trajectories from pluripotency in human neural organoid systems." Nature neuroscience 7(2024):27.
- Multiome 数据揭示 NEUROD2 基因染色质开放早于表达,结合多组学工具 pando 分析基因调控网络。 发现与神经发育相关的 NEUROD2 基因的表达在染色质开放时可能受到某种调控关系
 单细胞 Multiome 验证并得出 NEUROD2 基因上下游的调控关系
单细胞 Multiome 验证并得出 NEUROD2 基因上下游的调控关系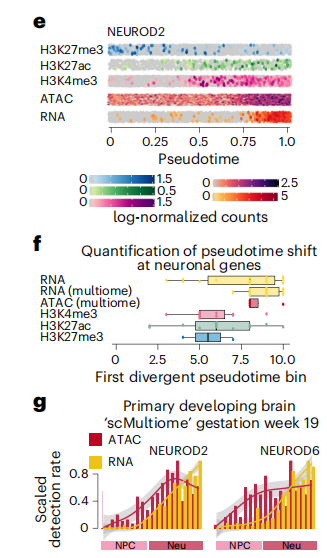
FAQ(常见问题)
- 如何选择单组学还是 Multiome? 若仅关注表达层面且预算有限,scRNA-seq 足够;若需同时解析调控与表达或构建基因调控网络,建议 Multiome(ATAC+RNA)。
- 质控阈值是否固定? 否。请以分布为依据,并结合项目目标微调。整体质量好时可上调 TSS 阈值以提升信噪比。
- TSS 富集低的常见原因? 核制备不佳、死细胞比例高、文库降解。建议复核核小体信号与 insert size 分布并严格过滤。
- 如何做可靠的标签转移? 选择高质量参考,匹配组织/物种,使用稳定 marker 面板并在多分辨率下交叉验证。
- 出现 RNA 与 ATAC 结果不一致时怎么办? 优先检查批次效应与峰/基因坐标一致性,辅以 motif/TF 活性与外部证据验证。
参考文献
- Hao, Yuhan, et al. "Integrated analysis of multimodal single-cell data." Cell 184.13(2021):3573-3587.e29.
- "Spatially resolved transcriptomic analysis of the adult human prostate." Nature Genetics.
- "Single-cell transcriptomic and chromatin dynamics of the human brain in PTSD." Nature.
- Trevino, Alexandro E., et al. "Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution." Cell D1(2021).
- Fides, Zenk, et al. "Single-cell epigenomic reconstruction of developmental trajectories from pluripotency in human neural organoid systems." Nature neuroscience 7(2024):27.
- Granja, Jeffrey M., et al. "Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia." Nature Biotechnology.
- Johain R. Ounadjela, Ke Zhang, et al. "Spatial multiomic landscape of the human placenta at molecular resolution." Nature Medicine.
- 云平台入口:SeekSoul Online