Datas
"Datas" lets you view the datasets under your account that can be used to create analysis pipelines, including SeekGene-associated standard analysis datasets and data uploaded by users.
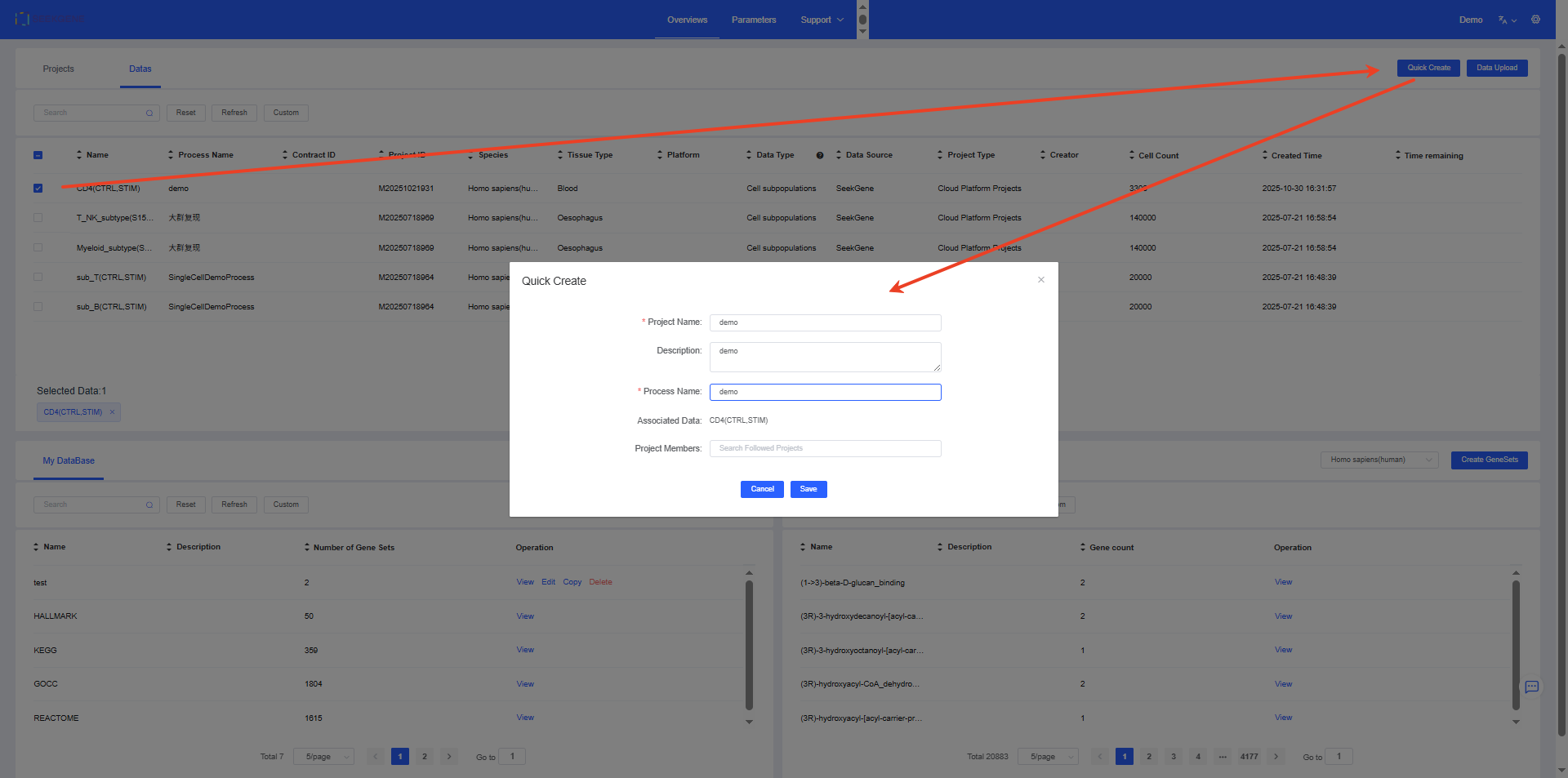
Quick Create
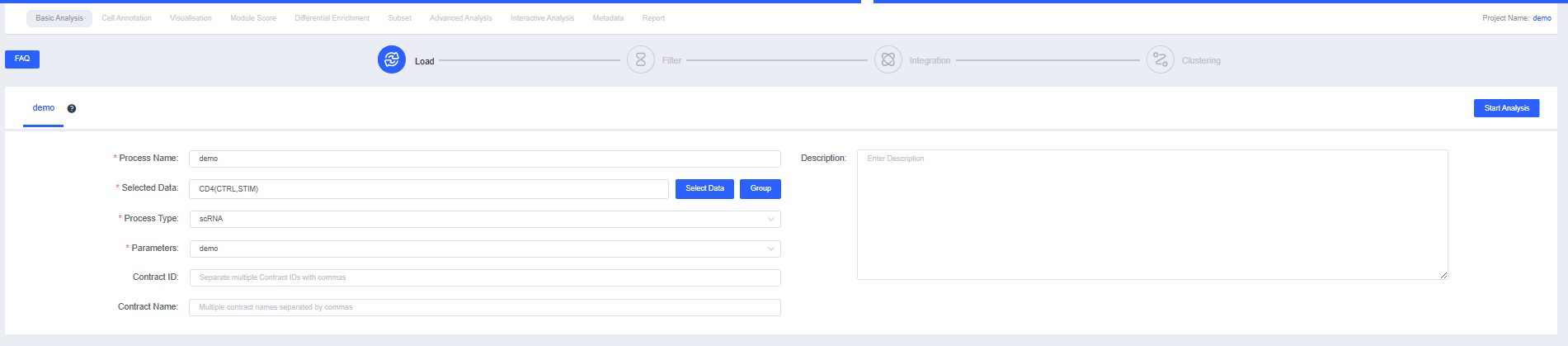
Select the sample data you want to analyze and click the [Quick Create] button to rapidly create a project and its analysis pipeline. After saving, you will be redirected to the new project pipeline for initialization. Subsequent steps can follow the earlier section on Basic Analysis.
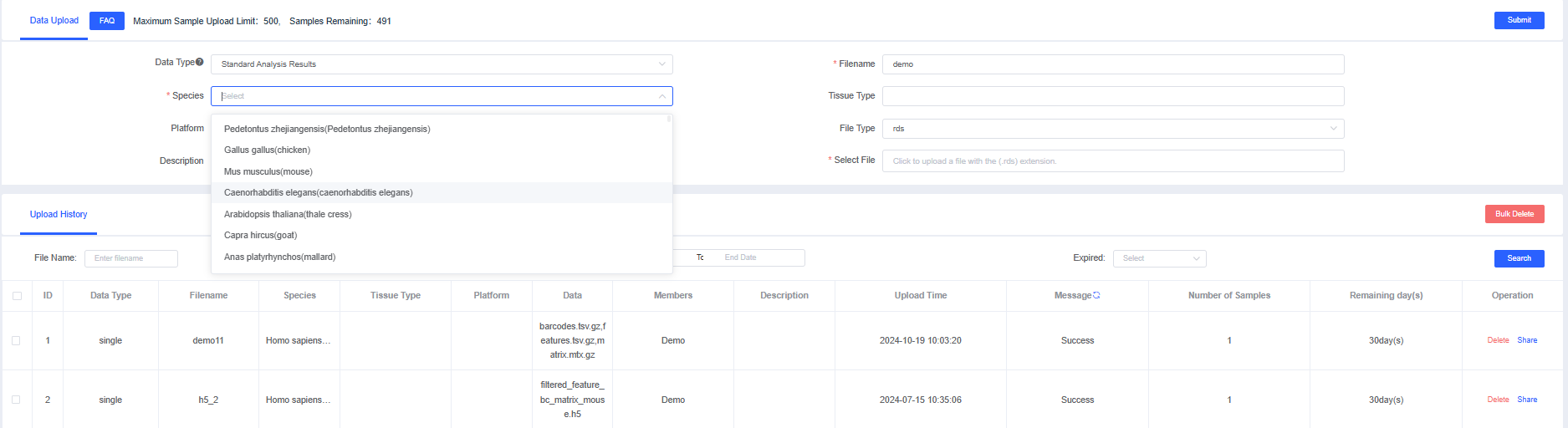
Data Upload
Data Upload is primarily used to upload single-cell datasets from your local machine for analysis. Click the button to open the Data Upload window, fill in the required information, and choose the data to upload. Data types are categorized into Standard Analysis Results and Single-sample Data.
Standard Analysis Results
Standard Analysis Results refer to files that have already completed single-cell standard analysis (filtering, normalization, scaling, dimensionality reduction/integration, clustering, etc.) using Seurat or scanpy. Only SeuratObject (rds) files are supported.
1.1 Upload an "rds" file of Standard Analysis Results, fill in the information, and click [Submit]. Once the validation status shows "Successful," you can create a pipeline for analysis and even [Share] the dataset with other users.
NOTE
Uploading Standard Analysis Results allows you to skip filtering and integration during Basic Analysis.
1.2 Formatting requirements for "rds" files classified as Standard Analysis Results:
- The data structure must be a SeuratObject (rds) object, the filename must end with .rds, and the metadata must include the labels nCount_RNA, nFeature_RNA, and RNA_snn_res.x.x (or resolution.x.x, where x.x is the resolution value, such as RNA_snn_res.0.5 or resolution.0.5).
- If no Sample label exists in the metadata, the uploaded filename is used as the sample name.
- If the metadata lacks percent.mito information, the proportion of genes beginning with MT-, mt-, or Mt- is automatically calculated and stored in a column named mito.
- A validation status of "Successful" indicates that the data upload succeeded.
| nCount_RNA (Required) | nFeature_RNA (Required) | RNA_snn_res.0.5 (Required) | Sample (Required for multisample) | percent.mt (Optional) | |
|---|---|---|---|---|---|
| AAACATACAACCAC-1 | 2419 | 779 | 4 | A | 3.0177759 |
| AAACATACAACTAC-1 | 4219 | 1727 | 3 | A | 0.8897363 |
| AAACATACAACGAC-1 | 980 | 545 | 2 | C | 1.2244898 |
| AAACATACAACCCC-1 | 263 | 132 | 4 | C | 1.6643551 |
1.3 How SeekSoulOnline reads "rds" files for Standard Analysis Results:
# Load package
library(Seurat)
# Read data
standard_demo_rds <- readRDS("standard_demo.rds")1.4 Common reasons why file validation fails:
- The Standard Analysis Results rds file is not a SeuratObject (rds).
- The metadata does not contain nCount_RNA or nFeature_RNA information.
- The metadata lacks the RNA_snn_res.x.x or resolution.x.x resolution labels.
- The metadata has no Sample label, so multisample data are treated as single-sample.
Single Sample Data
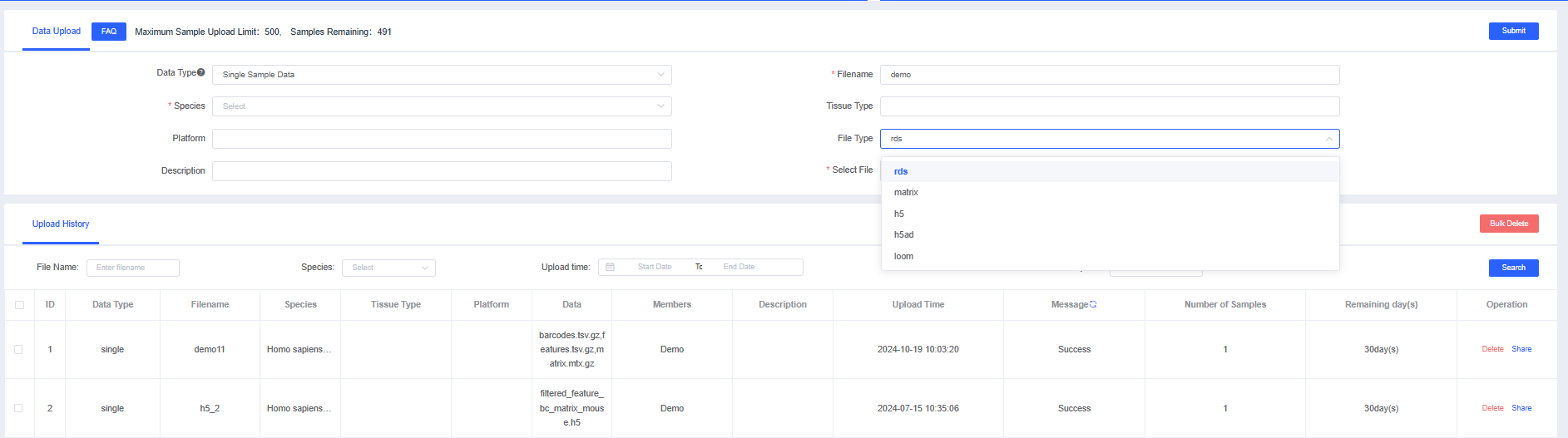
Single Sample Data refers to single-cell expression matrix files and supports SeuratObject (rds), data matrices (matrix), HDF5 (h5), Scanpy files (h5ad), and loom files based on HDF5.
2.1 Uploading Single-sample Data supports multiple formats, including "rds," "matrix," "h5," "h5ad," and "loom" files. Fill in the information, click [Submit], and once the validation status shows "Successful," you can create a pipeline for analysis and [Share] the dataset with other users.
2.2 How SeekSoulOnline reads "rds" files for Single Sample Data:
# Load package
library(Seurat)
# Read data
single_demo_rds <- readRDS("single_demo.rds")2.3 Formatting requirements for "matrix" files:
- Include barcodes.tsv (one column of barcodes), features.tsv (two columns for Ensembl ID and gene name), and matrix.mtx (header plus three columns for gene name, barcode, and counts).
barcodes.tsv example:
AAACATACAACCAC-1
AAACATTGAGCTAC-1
...features.tsv example:
ENSG00000243485 MIR1302-10
ENSG00000237613 FAM138A
...matrix.mtx example:
%%MatrixMarket matrix coordinate real general
%
32738 2700 2286884
32709 1 4
...2.4 How SeekSoulOnline reads "matrix" files:
# Load package
library(Seurat)
# Read data
dir = "./single_demo_matirx"
single_demo_matrix = CreateSeuratObject(Read10X(dir))2.5 How SeekSoulOnline reads "h5" files:
# Load package
library(Seurat)
# Read data
single_demo_h5 <- CreateSeuratObject(Read10X_h5("single_demo.h5"))2.6 How SeekSoulOnline reads "h5ad" files:
# Load package
library(SeuratDisk)
# Read data
Convert('single_demo.h5ad', "h5seurat", overwrite = TRUE, assay = "RNA")
single_demo_h5seurat <- LoadH5Seurat("single_demo.h5seurat")CAUTION
Columns in the h5ad data should be categorical variables; otherwise, running LoadH5Seurat raises the error "Missing required datasets 'levels' and 'values'", causing the upload to fail.
2.7 How SeekSoulOnline reads "loom" files:
# Load package
library(SeuratDisk)
# Read data
single_demo_loom <- LoadLoom("single_demo.loom")