Run
Usage
Activate Environment
conda activate seeksoulmethylRun Dual-omics Analysis (Shell script)
# sc_methy_workflow.sh can be found in the SeekSoulMethyl directory you cloned
bash sc_methy_workflow.sh \
/path/to/expression_R1.fastq.gz \
/path/to/expression_R2.fastq.gz \
/path/to/methy_R1.fastq.gz \
/path/to/methy_R2.fastq.gz \
--sample WTJW880 \
--outdir /path/to/results \
--database_dir /path/to/human-reference-GRCh38 \
--chemistry DD-MET3 \
--core 64 \
--filter_ch 2If a sample has multiple datasets, separate file paths with commas. Ensure that the FASTQ files are listed in the correct order for each dataset.
bash sc_methy_workflow.sh \
/path/to/WTJW969_E_L003_R1.fq.gz,/path/to/WTJW969_E_L004_R1.fq.gz \
/path/to/WTJW969_E_L003_R2.fq.gz,/path/to/WTJW969_E_L004_R2.fq.gz \
/path/to/WTJW969_Met_L000_R1.fq.gz,/path/to/WTJW969_Met_L001_R1.fq.gz,/path/to/WTJW969_Met_L002_R1.fq.gz,/path/to/WTJW969_Met_L003_R1.fq.gz,/path/to/WTJW969_Met_L004_R1.fq.gz \
/path/to/WTJW969_Met_L000_R2.fq.gz,/path/to/WTJW969_Met_L001_R2.fq.gz,/path/to/WTJW969_Met_L002_R2.fq.gz,/path/to/WTJW969_Met_L003_R2.fq.gz,/path/to/WTJW969_Met_L004_R2.fq.gz \
--sample WTJW969 \
--outdir /path/to/results \
--database_dir /path/to/human-reference-GRCh38 \
--chemistry DD-MET5 \
--core 64 \
--filter_ch 2Input Parameters:
- $1: Single-cell transcriptome Read1 fastq file path.
- $2: Single-cell transcriptome Read2 fastq file path.
- $3: Single-cell methylation Read1 fastq file path.
- $4: Single-cell methylation Read2 fastq file path.
- sample: Sample name.
- outdir: Output directory path.
- database_dir: Reference genome database path.
- chemistry: Methylation chemistry (DD-MET3 or DD-MET5). DD-MET3 is a dual-label dataset, meaning the RNA and DNA methylation data barcodes are different for the same cell, while DD-MET5 is single-label, meaning the RNA and DNA methylation data barcodes are the same for the same cell.
- core: Number of CPU cores.
- filter_ch: Filter reads that contain > n CH methylation sites. If you do not want to enable this filter, set filter_ch to 0.
Quick Start with Nextflow (Recommended)
If you want to process samples in batch and get pipeline-level reports, use Nextflow:
conda activate seeksoulmethyl
conda install -n seeksoulmethyl -c bioconda nextflow
# Prepare the sample sheet (see example above)
nextflow run SeekSoulMethyl/nf/main.nf \
--outdir /path/to/results \
--samplesheet samplelist.csv \
-w /path/to/work \
-c SeekSoulMethyl/nf/nextflow.config \
-profile aliyun_k8s \
--database_dir /path/to/reference \
--split_fastq 4 \
--filter_ch 2 \
--chemistry DD-MET3 > methy.log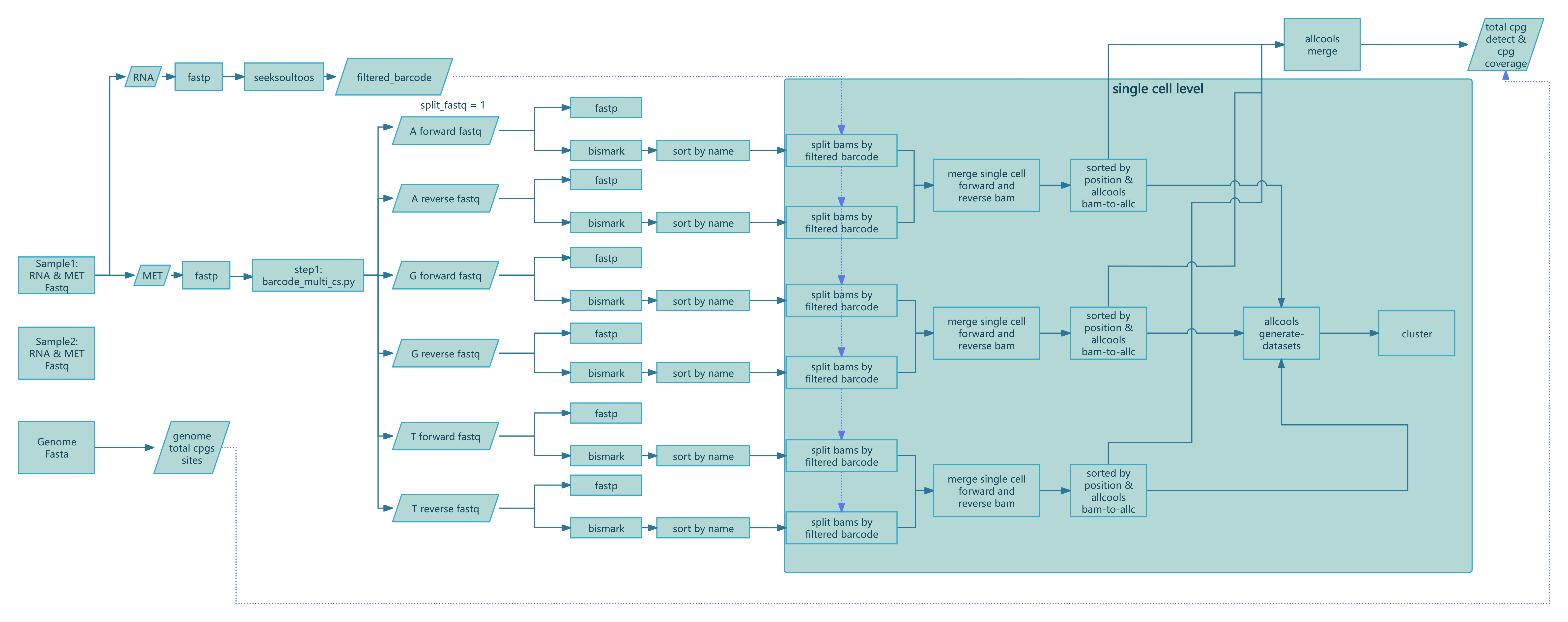
Running with Nextflow
- Install nextflow:
conda install -n seeksoulmethyl -c bioconda nextflow- Prepare your input samplesheet
cat > samplelist.csv << EOF
sample_id,expression_r1,expression_r2,methylation_r1,methylation_r2
XYRD-WTJW880,/path/to/XYRD-WTJW880-E_S1_L005_R1_001.fastq.gz,/path/to/XYRD-WTJW880-E_S1_L005_R2_001.fastq.gz,/path/to/XYRD-WTJW880-MET_S01_L001_R1_001.fastq.gz,/path/to/XYRD-WTJW880-MET_S01_L001_R2_001.fastq.gz
EOF
# expression_r1: Single-cell transcriptome Read1 fastq file
# expression_r2: Single-cell transcriptome Read2 fastq file
# methylation_r1: Single-cell methylation Read1 fastq file
# methylation_r2: Single-cell methylation Read2 fastq fileIf a single sample has multiple datasets and the transcriptome and methylation FASTQ counts do not match (e.g., WTJW969), add multiple rows to the samplesheet, with each row representing one dataset.
sample_id,expression_r1,expression_r2,methylation_r1,methylation_r2
WTJW969,/path/to/WTJW969_E_L003_R1.fq.gz,/path/to/WTJW969_E_L003_R2.fq.gz,/path/to/WTJW969_Met_L000_R1.fq.gz,/path/to/WTJW969_Met_L000_R2.fq.gz
WTJW969,/path/to/WTJW969_E_L004_R1.fq.gz,/path/to/WTJW969_E_L004_R2.fq.gz,/path/to/WTJW969_Met_L001_R1.fq.gz,/path/to/WTJW969_Met_L001_R2.fq.gz
WTJW969,,,/path/to/WTJW969_Met_L002_R1.fq.gz,/path/to/WTJW969_Met_L002_R2.fq.gz
WTJW969,,,/path/to/WTJW969_Met_L003_R1.fq.gz,/path/to/WTJW969_Met_L003_R2.fq.gz
WTJW969,,,/path/to/WTJW969_Met_L004_R1.fq.gz,/path/to/WTJW969_Met_L004_R2.fq.gz- Run the pipeline:
nextflow run -bg SeekSoulMethyl/nf/main.nf \
--outdir /path/to/tiny_demo/results/ \
--samplesheet samplelist.csv \
-w /path/to/tiny_demo/results/work \
-c SeekSoulMethyl/nf/nextflow.config \
-profile aliyun_k8s \
--database_dir /path/to/human-reference-GRCh38/ \
--split_fastq 1 \
--filter_ch 2 \
--chemistry DD-MET3 > methy.log
# --outdir: final results directory
# --samplesheet: input samplesheet file
# -w: working directory for nextflow
# -c: nextflow configuration file. Must be modified according to your server configuration.
# -profile: nextflow profile for aliyun k8s
# --database_dir: reference genome database directory
# --split_fastq: To speed up the analysis process, split fastq according to first n bases of barcode. Default is 4.
# --filter_ch: filter reads that contain > 2 CH methylation sites.
# --chemistry: methylation chemistry (DD-MET3 or DD-MET5)Notes on nextflow.config
nextflow.config declares the execution environment, resource quotas, and run policies. You must customize it to your own server or cluster.
- Location:
SeekSoulMethyl/nf/nextflow.config(specify with-c SeekSoulMethyl/nf/nextflow.configwhen running). - Key items to tailor to your infrastructure:
- Executor:
process.executor(e.g.,local,slurm,pbs,k8s,awsbatch). - Resources:
process.cpus,process.memory,process.time, or fine-grained overrides viawithLabel/withName. - Work directory:
workDir(can also be set via-w); ensure it is writable and has sufficient space. - Environment: enable one of
conda.enabled,docker.enabled, orsingularity.enabledaccording to what your server supports.
- Executor:
Example configurations (replace paths and parameters with values valid on your servers):
profiles {
// Local machine
local {
process.executor = 'local'
workDir = '/path/to/work'
process.cpus = 8
process.memory = '32 GB'
conda.enabled = true
// Or containers: singularity.enabled = true / docker.enabled = true
}
// Slurm cluster (HPC)
slurm {
process.executor = 'slurm'
workDir = '/lustre/work/your_user'
process.cpus = 8
process.memory = '32 GB'
process.queue = 'normal'
process.clusterOptions = '-A your_account --qos=normal'
withLabel: 'high_mem' {
cpus = 16
memory = '64 GB'
}
}
// Kubernetes (e.g., Alibaba Cloud ACK)
aliyun_k8s {
process.executor = 'k8s'
workDir = '/mnt/nf-work' // persistent volume path
k8s {
namespace = 'your-namespace'
storageClaimName = 'your-pvc'
cpu = 4
memory = '16 GB'
}
// If using container images globally: docker.enabled = true
}
}Tips:
- Pick the
-profilethat matches your environment (e.g.,slurm,local,aliyun_k8s), then adapt the parameters. - Keep
workDiron fast storage with ample capacity; the final results directory is controlled by--outdir. - If you use the README’s conda environment, prefer
conda.enabled; if your cluster favors containers, use Docker/Singularity.
References:
- Nextflow configuration & profiles: https://www.nextflow.io/docs/latest/config.html
- Executors (local, Slurm, K8s, etc.): https://www.nextflow.io/docs/latest/executor.html
- Kubernetes guide: https://www.nextflow.io/docs/latest/kubernetes.html
Nextflow Step-by-Step Details
This section describes each Nextflow process with inputs, core logic, key parameters, and outputs for troubleshooting and interpretation. The workflow is defined in nf/main.nf (script/SeekSoulMethyl/nf/main.nf:1) and processes are implemented in nf/modules/*.nf.
Step 1: Preprocessing and Barcode Parsing (step1.nf)
Compute genome-wide CpG count:
COMPUTE_CPG_SITES(script/SeekSoulMethyl/nf/modules/step1.nf:6)- Input:
params.genomefa,params.chrom_size_path - Action: run
count_cg_sites.pyto count CpG sites - Output:
genome_cg_info.json
- Input:
Expression FASTQ QC (multi-group):
FASTP_EXPRESSION_MULTI(script/SeekSoulMethyl/nf/modules/step1.nf:22)- Input: paired FASTQs per sample (groups G1/G2/...)
- Action:
fastptrimming and QC - Output: cleaned
*_expression_clean_R1/2.fastq.gz,*.html,*.json
Methylation FASTQ QC (multi-group):
FASTP_METHYLATION_MULTI(script/SeekSoulMethyl/nf/modules/step1.nf:88)- Input: paired FASTQs per sample (groups G1/G2/...)
- Action:
fastpQC (adapter detection disabled, trimming as per pipeline) - Output: cleaned
*_methylation_clean_R1/2.fastq.gz,*.html,*.json
RNA alignment and quantification:
SEEKSOULTOOLS_RNA(script/SeekSoulMethyl/nf/modules/step1.nf:156)- Input: cleaned expression R1/R2 lists
- Action:
seeksoultools rna run(STAR mapping, counting, filtering, clustering, DE) - Output:
Analysis/step3/filtered_feature_bc_matrix/etc.
Methylation barcode parsing:
METHYLATION_BARCODE_EXTRACTION(script/SeekSoulMethyl/nf/modules/step1.nf:206)- Input: cleaned methylation R1/R2 lists, whitelist
params.methy_barcode_wl - Action: run
barcode_cs_multi.pyto parse/correct cell barcodes and UMIs byparams.chemistry; optional--split_fastq nto shard reads by first n barcode bases - Output:
step1/${sample}_forward_*{1,2}.fq.gz,step1/${sample}_reverse_*{1,2}.fq.gz,${sample}_methy_summary.json, optional${sample}_barcode_stats.txt
- Input: cleaned methylation R1/R2 lists, whitelist
Build forward/reverse pairing lists:
PARSE_FASTQ_FILES(script/SeekSoulMethyl/nf/modules/step1.nf:255)- Input: forward/reverse FASTQ sets
- Action: scan
step1/and pair files by identifiers - Output:
forward_pairs.txt,reverse_pairs.txt
Post-barcode-extraction QC:
FASTP_METHYLATION_BARCODE_EXTRACT(script/SeekSoulMethyl/nf/modules/step1.nf:368)- Input: paired sub-FASTQs
- Action:
fastpQC - Output: per-pair
*.html,*.json
Step 2: Bismark alignment and BAM sorting (step2.nf)
Forward-strand alignment:
BISMARK_ALIGNMENT_FORWARD(script/SeekSoulMethyl/nf/modules/step2.nf:3)- Key flags:
--add_barcode,--add_umi; max insert size-X 1000 - Output:
*_bismark_bt2_pe.bam,*_bismark_bt2_PE_report.txt
- Key flags:
Reverse (PBAT) alignment:
BISMARK_ALIGNMENT_REVERSE(script/SeekSoulMethyl/nf/modules/step2.nf:31)- Key flags:
--pbat,--add_barcode,--add_umi - Output: same as above
- Key flags:
Sort by read name:
SORT_BAM_BY_NAME(script/SeekSoulMethyl/nf/modules/step2.nf:61)- Action:
samtools sort -n - Output:
*_sortbyname.bam
- Action:
Step 3: Per-cell split, ALLC generation/merge, dataset building (step3.nf)
Split BAMs by cell barcode:
SPLIT_BAM_FILES(script/SeekSoulMethyl/nf/modules/step3.nf:1)- Input: name-sorted BAM and GEX barcodes
barcodes.tsv.gz - Action: run
step3_split_bams.pyto split by cell barcode and keep shared cells only - Output: per-cell BAM dir,
*_filtered_barcode,*_filtered_barcode_reads_counts.csv
- Input: name-sorted BAM and GEX barcodes
Merge forward/reverse per-cell BAMs and counts:
MERGE_BISMARK_BAM(script/SeekSoulMethyl/nf/modules/step3.nf:27)- Action:
samtools merge -nper matching cell; merge/deduplicate barcodes and read counts - Output:
*_merged_fr_bam/,*_merge_filtered_barcode,*_merge_filtered_barcode_reads_counts.csv
- Action:
Generate per-cell ALLC:
ALLCOOLS_BAM_TO_ALLC(script/SeekSoulMethyl/nf/modules/step3.nf:86)- Action: run
step3_bam_to_allc.py(custom ALLCools), UR-based dedup and methylation quantification - Output: per-cell
*_allc.gzand index
- Action: run
Merge cell metrics:
MERGE_FILTERED_BARCODE_READS_COUNTS(script/SeekSoulMethyl/nf/modules/step3.nf:114)- Action: merge barcodes and read counts, create
${sample}_cells.csvand.json - Output:
filtered_barcode,filtered_barcode_reads_counts.csv,${sample}_cells.{csv,json}
- Action: merge barcodes and read counts, create
Build multi-scale dataset:
ALLCOOLS_GENERATE_DATASETS(script/SeekSoulMethyl/nf/modules/step3.nf:136)- Action:
allcools generate-datasetfor chrom10k/20k/50k/100k/500k/1M, metrics likecountandhypo-score - Output:
${sample}.mcds
- Action:
Merge ALLC (when sharded):
ALLCOOLS_SUBMERGE(script/SeekSoulMethyl/nf/modules/step3.nf:188),ALLCOOLS_MERGE(script/SeekSoulMethyl/nf/modules/step3.nf:223)- Action: merge per-shard/per-sample ALLCs
- Output:
${sample}_merge_allc.gzand index
Extract CG context ALLC:
ALLCOOLS_EXTRACT(script/SeekSoulMethyl/nf/modules/step3.nf:252)- Action:
allcools extract-allc --mc_contexts CGN --strandness merge - Output:
*.CGN-Merge*
- Action:
(Methylation-only workflow methy_only.nf additionally includes COUNTS_MAPPED_READS and ESTIMATED_CELLS for read-count-based cell estimation and barcode filtering, see script/SeekSoulMethyl/nf/modules/utils.nf:1 and :17)
Step 4: Summary, DR & joint report (step4.nf)
Methylation summary:
METHYLATION_SUMMARY(script/SeekSoulMethyl/nf/modules/step4.nf:2)- Action:
step4_wgs_summary.pyaggregates Bismark reports, cell metrics and CpG stats to produce${sample}_methy_summary.jsonand${sample}_wgs_summary.csv
- Action:
LSI/PCA clustering and visualization:
METHYLATION_LSI_PCA_CLUSTERING(script/SeekSoulMethyl/nf/modules/step4.nf:26)- Action:
step4_allcools_PCA_cluster.py --var_dim chrom20k --reduc lsi - Output:
*.h5ad,*.pdf,*.png
- Action:
Transcriptome+Methylation joint report:
MULTI_REPORT(script/SeekSoulMethyl/nf/modules/step4.nf:52)- Action:
step4_report_rna_met.pyintegrates transcriptome and methylation outputs - Output:
${sample}_rna_methyl_report.html,${sample}_rna_met.json
- Action:
Methylation-only workflow (test version, currently not recommended for use)
Use the simplified workflow when you only have methylation reads:
nextflow run SeekSoulMethyl/nf/methy_only.nf \
--outdir /path/to/results \
--samplesheet samplelist.csv \
-w /path/to/work \
-c SeekSoulMethyl/nf/nextflow.config \
-profile aliyun_k8s \
--database_dir /path/to/reference \
--split_fastq 4 \
--filter_ch 2 \
--chemistry DD-MET3Key parameters and tips
--database_dir: reference directory withfasta/genome.fa,genes/genes.gtf,star/,bed/chr_nochrM.bed(script/SeekSoulMethyl/nf/main.nf:19)--chemistry:DD-MET3orDD-MET5; also sets transcriptome chemistry and barcode whitelist (script/SeekSoulMethyl/nf/main.nf:27)--split_fastq: shard by the first n barcode bases (default 4; increases parallelism but adds scheduling/merge overhead) (script/SeekSoulMethyl/nf/main.nf:36)--filter_ch: filter reads with > n CH methylation sites (default 2) (script/SeekSoulMethyl/nf/modules/step1.nf:241)- The samplesheet header must be
sample_id(script/SeekSoulMethyl/nf/main.nf:116)
Execution environment and resources
- Recommended container image:
registry.cn-beijing.aliyuncs.com/seekgene/seeksoulmethyl:1.1.2(script/SeekSoulMethyl/nf/nextflow.config:69) - Choose
-profile aliyun_k8sor-profile docker, and editnf/nextflow.configfor your infra - Heavy steps: Bismark/ALLCools need substantial CPU/RAM; defaults are set in
withNameblocks, scale up if needed (script/SeekSoulMethyl/nf/nextflow.config:313)
System Requirements
If you use sc_methy_workflow.sh, the system requirements are as follows:
- CPU: 64 cores (recommended)
- Memory: 128GB RAM (recommended)
- Storage: At least 500GB available space
- OS: Linux (recommended Ubuntu 18.04+ or CentOS 7+)
FAQ
- Samplesheet parsing error: ensure first column is
sample_id, use absolute paths (script/SeekSoulMethyl/nf/main.nf:111) - Missing
${sample}.mcds: checkALLCOOLS_BAM_TO_ALLCproduced per-cell*_allc.gzandchrom_size_pathis correct - Stuck at Bismark: verify reference indices and that
params.bismark_refis visible in the container - Resume runs: use
-resumewith the same-wwork directory