Algorithm
Data Structure
SeekOne DD Single Cell Multiome Methylation + RNA kit comes in two chemistries. DD-MET3 (dual-label) means the RNA and DNA methylation data barcodes are different for the same cell, and the RNA library is a 3′-end transcriptome library. DD-MET5 (single-label) means the RNA and DNA methylation data barcodes are the same for the same cell, and the RNA library is a 5′-end transcriptome library. Below we describe the DNA methylation library structures for both chemistries.
DD-MET3 Methylation Library Structure
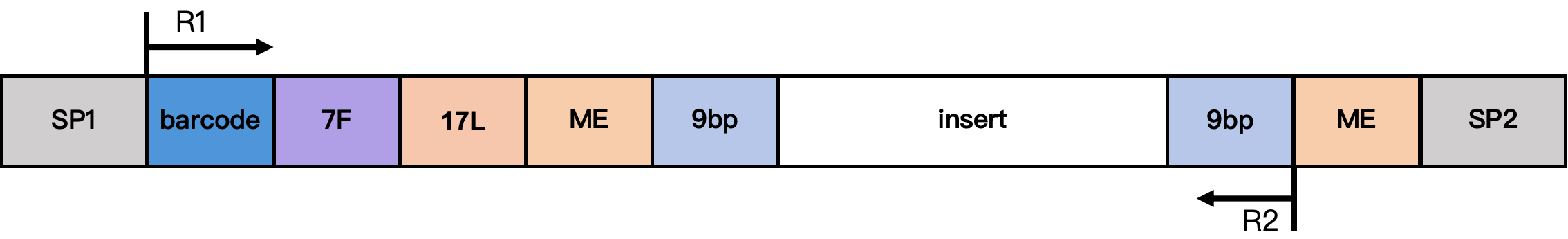
Structure notes:
- SP1/SP2: Adapter sequences
- barcode: 17 bp cell barcode
- 7F: 7 bp linker sequence
- 17L: 17 bp fixed sequence CgtCCgtCgttgCtCgt
- ME: 19 bp fixed sequence AGATGTGTATAAGAGACAG
- 9 bp: extension sequence from the Tn5 insertion fragment
DD-MET5 Methylation Library Structure
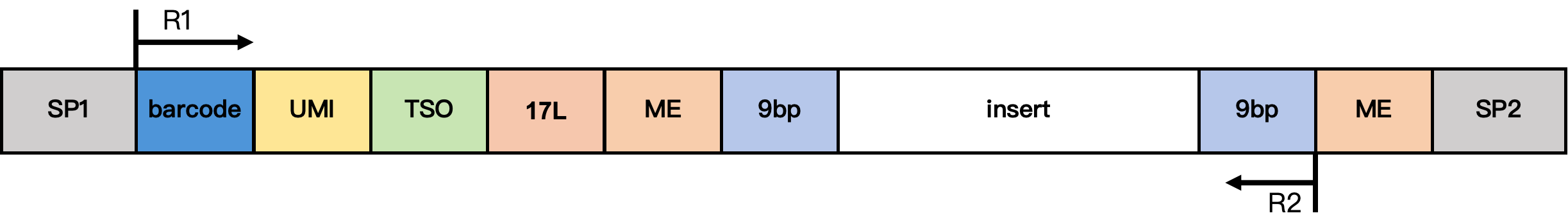
Structure notes:
- SP1/SP2: Adapter sequences
- barcode: 17 bp cell barcode
- UMI: 12 bp UMI sequence
- TSO: 13 bp TSO sequence TTTCTTATATGGG
- 17L: 17 bp fixed sequence CgtCCgtCgttgCtCgt
- ME: 19 bp fixed sequence AGATGTGTATAAGAGACAG
- 9 bp: extension sequence from the Tn5 insertion fragment
Since the enzymatic treatment converts unmethylated cytosines (C) to thymines (T), the C bases in SP1 and SP2 are methylated to prevent errors in the sequencing adapters during this conversion. Furthermore, the barcodes used for methylation data do not contain any C bases. In contrast, the C bases in 7F, 17L, and ME are not methylated and will be converted to T during the enzymatic process; we use these fixed sequences to calculate the C-to-T conversion rate.
Transcriptome Processing Workflow
Transcriptome data is analyzed using SeekSoul™ Tools. See the official Algorithms Overview for detailed steps. Cells used in the downstream methylation library are determined based on the transcriptome library cell barcodes.
Methylation Processing Workflow
Step 1: Preprocessing and Barcode Parsing
Barcode extraction and correction
Based on the designed structure, we locate the barcode in the read and extract the corresponding sequence. If the extracted barcode is in the whitelist, it is counted as a valid barcode; otherwise, SeekSoul™ Methyl Tools attempts barcode correction, if the barcode has a one-base mismatch (Hamming distance = 1) from an entry in the whitelist:
- If exactly one whitelist candidate matches: correct the invalid barcode to that whitelist barcode.
- If multiple whitelist candidates match: correct to the candidate supported by the highest read count.
If correction fails, the read is discarded and considered a final invalid barcode read.
UMI extraction
UMI positions are read from the designed structure and extracted without correction.
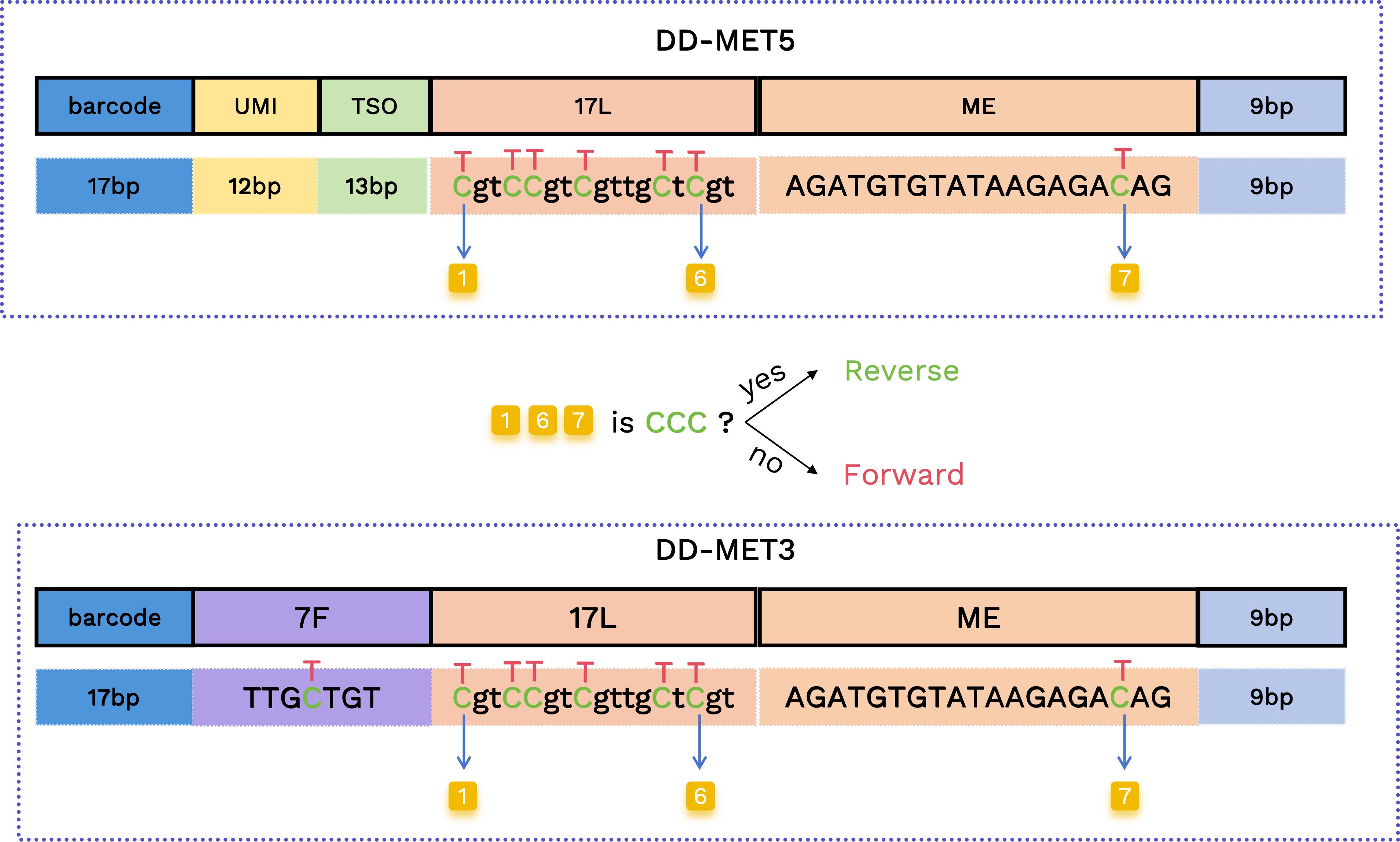
Forward and Reverse reads determination
From the positions corresponding to 17L and ME, there are 7 bases that can be C or converted T (highlighted below). We use the first and the last two C/T positions to determine forward vs. reverse reads: if all three positions are C, it indicates a reverse read; otherwise, it is a forward read.
Forward: TgtTTgtTgttgTtTgtAGATGTGTATAAGAGAT
Reverse: CgtCCgtCgttgCtCgtAGATGTGTATAAGAGAC
Reverse reads correspond to CTOT/CTOB (reverse complement of the original strand) in methylation terminology; forward reads correspond to OT/OB (original strand). The "forward" or "reverse" determination is annotated in the read name.
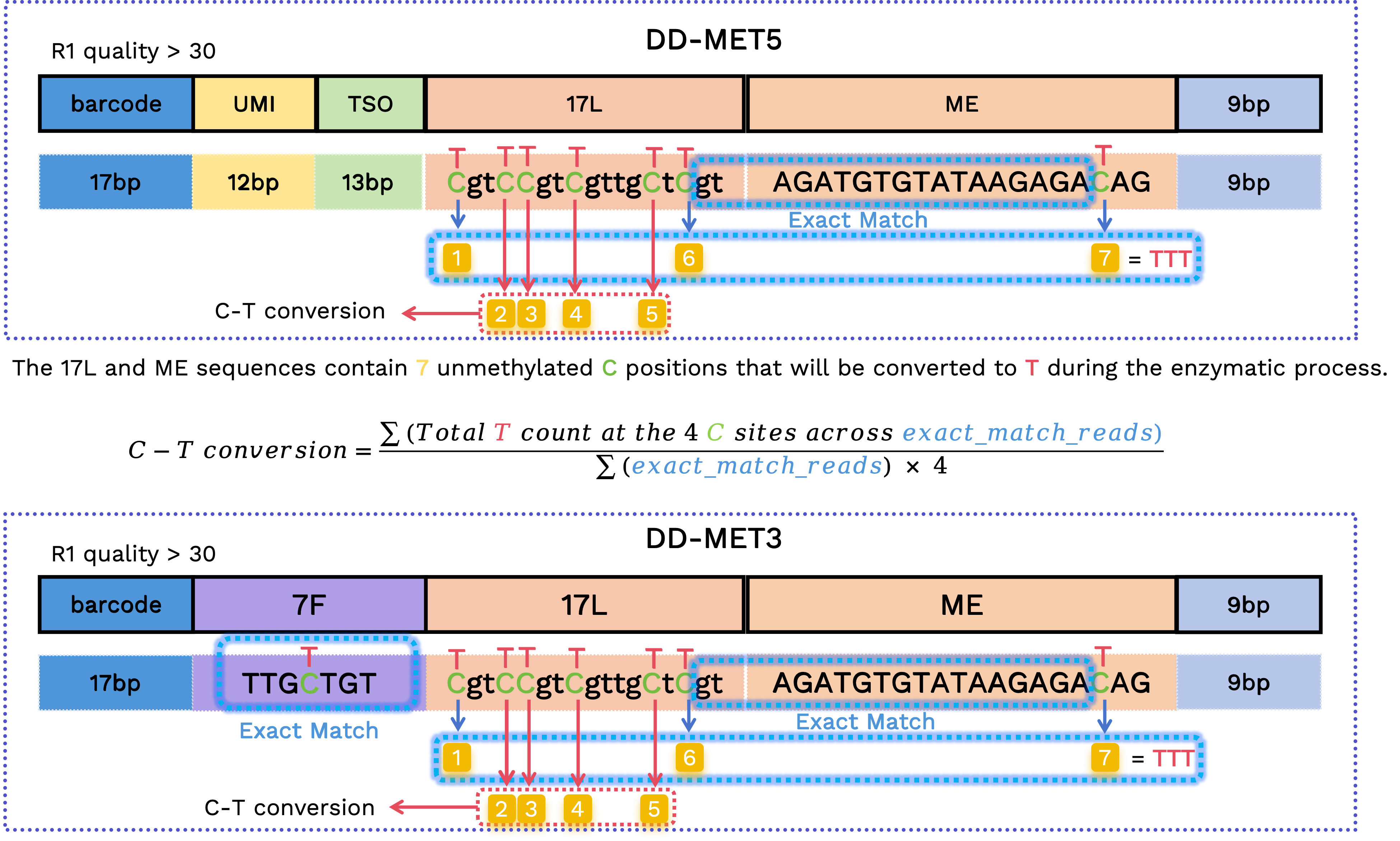
C–T conversion rate
We calculate the C-to-T conversion rate using the original C positions within 17L and ME sequences. Since these are fixed sequences prone to sequencing errors, we restrict the calculation to reads with verified structures:
In DD-MET3, the 7F sequence must be
TTGCTGTorTTGTTGT, the sequence spanning 17L and ME must beGTAGATGTGTATAAGAGA, and the bases at first and the last two original C positions must be T.In DD-MET5, the sequence spanning 17L and ME must be
GTAGATGTGTATAAGAGA, and the bases at the first and last two original C positions must be T.
For the retained reads, we extract the bases at the corresponding positions to calculate the C-to-T conversion rate:
NOTE
The above filtering steps are used only for calculating the C-to-T conversion rate; reads that do not meet these criteria are not filtered out from the final output FASTQ files.
Artifact removal
Remove TSO/7F linker, 17L and ME sequences from Read1 according to their predefined positions in the library design.
Adapter trimming
Use cutadapt to remove ME adapter sequences introduced by R1/R2 read-through events (overlapping paired-end reads).
Trim 9 bp gaps introduced by Tn5 transposase
After removing adapters and other artificial sequences, we additionally trim the 9 bp gaps flanking the inserted fragment that are introduced by Tn5 transposition. These 9 bp regions can carry artificial methylation and spuriously elevate CH methylation adjacent to the insert boundaries, so they are removed prior to downstream analysis.
Filter reads with too many non-CpG methylated C bases (optional)
Filter based on the number of non-CpG methylated C bases in a read pair. By default, pairs with > 2 non-CpG methylated Cs detected in read1/read2 are removed. If you do not want to enable this filter, set filter_ch to 0.
NOTE
This filtering strategy is based on findings by Lu et al. [1], which suggest that nicks in synthesized adapters can trigger Bst polymerase nick translation. This activity incorporates 5-methyl-dCTPs, leading to completely unconverted reads that appear as artificial methylation signals.
Filter too short reads After the preceding filtering and adapter trimming steps, if the length of R1 in a read pair is less than 20 bp or the length of R2 is less than 60 bp, the read pair is filtered out.
Step 2: Bismark alignment and sorting by name
Alignment and tagging
We use Bismark for methylation alignment. Our modified Bismark adds --add_barcode and --add_umi to tag BAM files by read name with CB (error-corrected barcode) and UR (raw UMI). For forward reads, we use -X 1000 to allow insert sizes up to 1000 bp; for reverse reads, we use --pbat and -X 1000. After alignment, sort BAMs by read name using samtools sort -n; the name-sorted BAMs serve as inputs for downstream analysis.
Step 3: ALLCools analysis
Split by cell barcode
Split name-sorted BAMs by RNA-derived cell barcodes into per-cell BAM files, each containing uniquely mapped reads for one cell.
Generate ALLC files
Sort each per-cell BAM by position and convert to ALLC using ALLCools bam-to-allc. Our modified ALLCools performs UR-tag-based UMI correction and deduplication per C site.
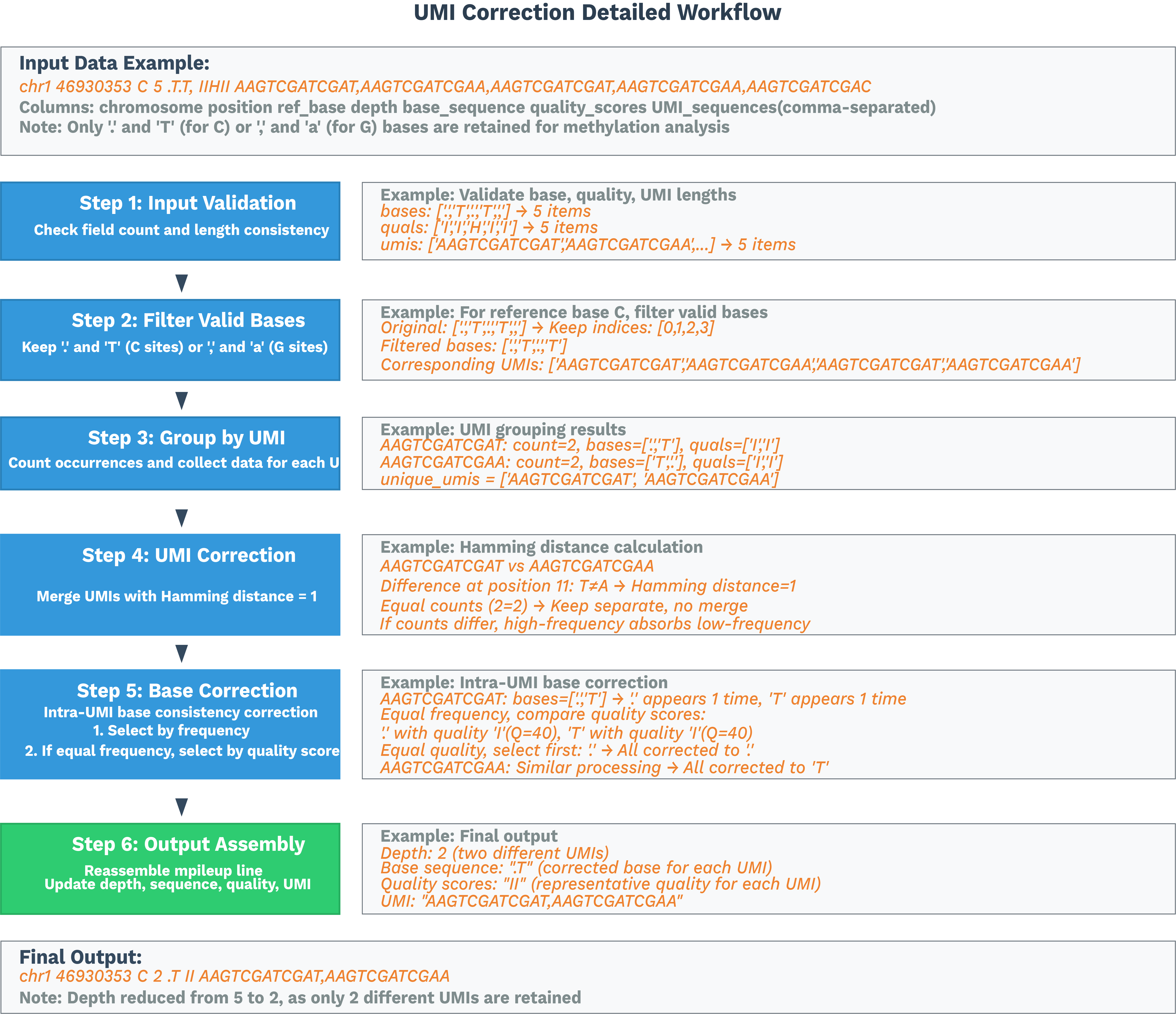
See the SeekGene ALLCools repository for details.
Generate MCDS
Run allcools generate-dataset to bin the genome (chrom10k/20k/50k/100k/500k/1M/geneslop2k) and compute per-cell methylation matrices. Geneslop2k bins are defined as 2k bp flanking each gene.
Step 4: Reduction and clustering
By default, perform dimensionality reduction with LSI on chrom20k bins using ALLCools, followed by UMAP visualization.