算法原理
数据结构
SeekOne DD Single Cell Multiome Methylation + RNA 试剂盒提供两种版本。 DD-MET3(双标签)表示同一细胞的 RNA 和 DNA 甲基化数据具有不同的条形码 (Barcodes),且 RNA 文库为 3' 端转录组文库。 DD-MET5(单标签)表示同一细胞的 RNA 和 DNA 甲基化数据具有相同的条形码,且 RNA 文库为 5' 端转录组文库。下文将详细介绍这两种化学体系的 DNA 甲基化文库结构。
DD-MET3 甲基化文库结构
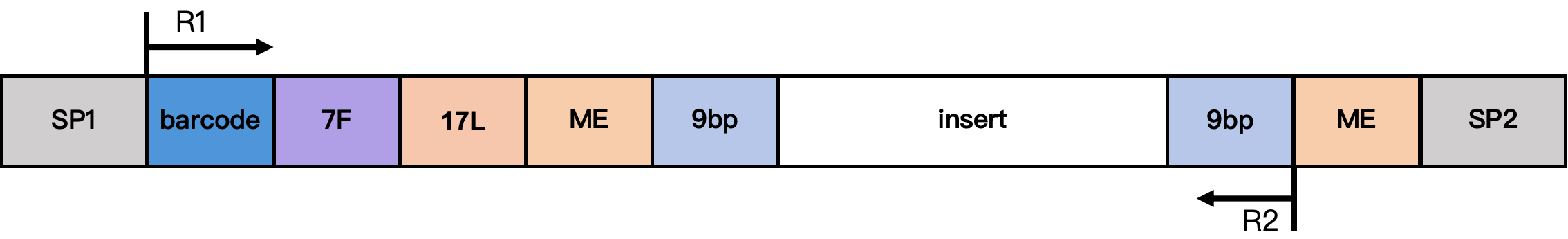
结构说明:
- SP1/SP2:接头序列
- barcode:17 bp 细胞 Barcode
- 7F:7 bp 连接序列
- 17L:17 bp 固定序列 CgtCCgtCgttgCtCgt
- ME:19 bp 固定序列 AGATGTGTATAAGAGACAG
- 9 bp:来自 Tn5 插入片段的延伸序列
DD-MET5 甲基化文库结构
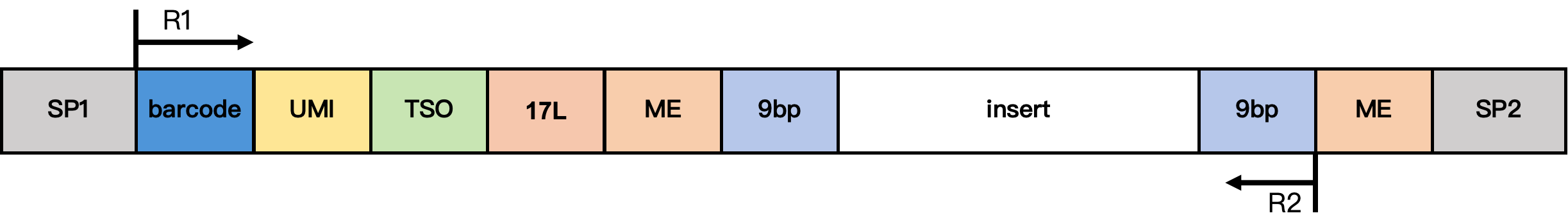
结构说明:
- SP1/SP2:接头序列
- barcode:17 bp 细胞barcode
- UMI:12 bp UMI 序列
- TSO:13 bp TSO 序列 TTTCTTATATGGG
- 17L:17 bp 固定序列 CgtCCgtCgttgCtCgt
- ME:19 bp 固定序列 AGATGTGTATAAGAGACAG
- 9 bp:来自 Tn5 插入片段的延伸序列
由于酶处理会将未甲基化的胞嘧啶(C)转换为胸腺嘧啶(T),因此 SP1 和 SP2 中的 C 碱基经过甲基化修饰,以防止测序接头在此转换过程中发生错误。此外,用于甲基化数据的条形码不包含任何 C 碱基。相比之下,7F、17L 和 ME 中的 C 碱基未甲基化,将在酶处理过程中转换为 T;我们利用这些固定序列来计算 C 到 T 的转换率。
转录组处理流程
转录组数据使用 SeekSoul Tools 进行分析。详细步骤请参考官方 算法概述。下游甲基化文库中使用的细胞是根据转录组文库的细胞条形码确定的。
甲基化处理流程
步骤 1:预处理和条形码解析
条形码提取和校正
根据设计的结构,我们在 Reads 中定位条形码并提取相应的序列。如果提取的条形码在白名单中,则视为有效条形码;若不在白名单中,但与白名单条目的汉明距离为 1(即存在一个碱基不匹配),SeekSoul Methyl Tools 将尝试进行条形码校正:
- 如果只有一个白名单候选者匹配:将无效条形码校正为该白名单条形码。
- 如果有多个白名单候选者匹配:校正为 Reads 计数最高的候选者。
如果校正失败,该 Reads 将被丢弃,并被视为最终的无效条形码 Read。
UMI 提取
从设计的结构中读取 UMI 位置并提取,不进行校正。
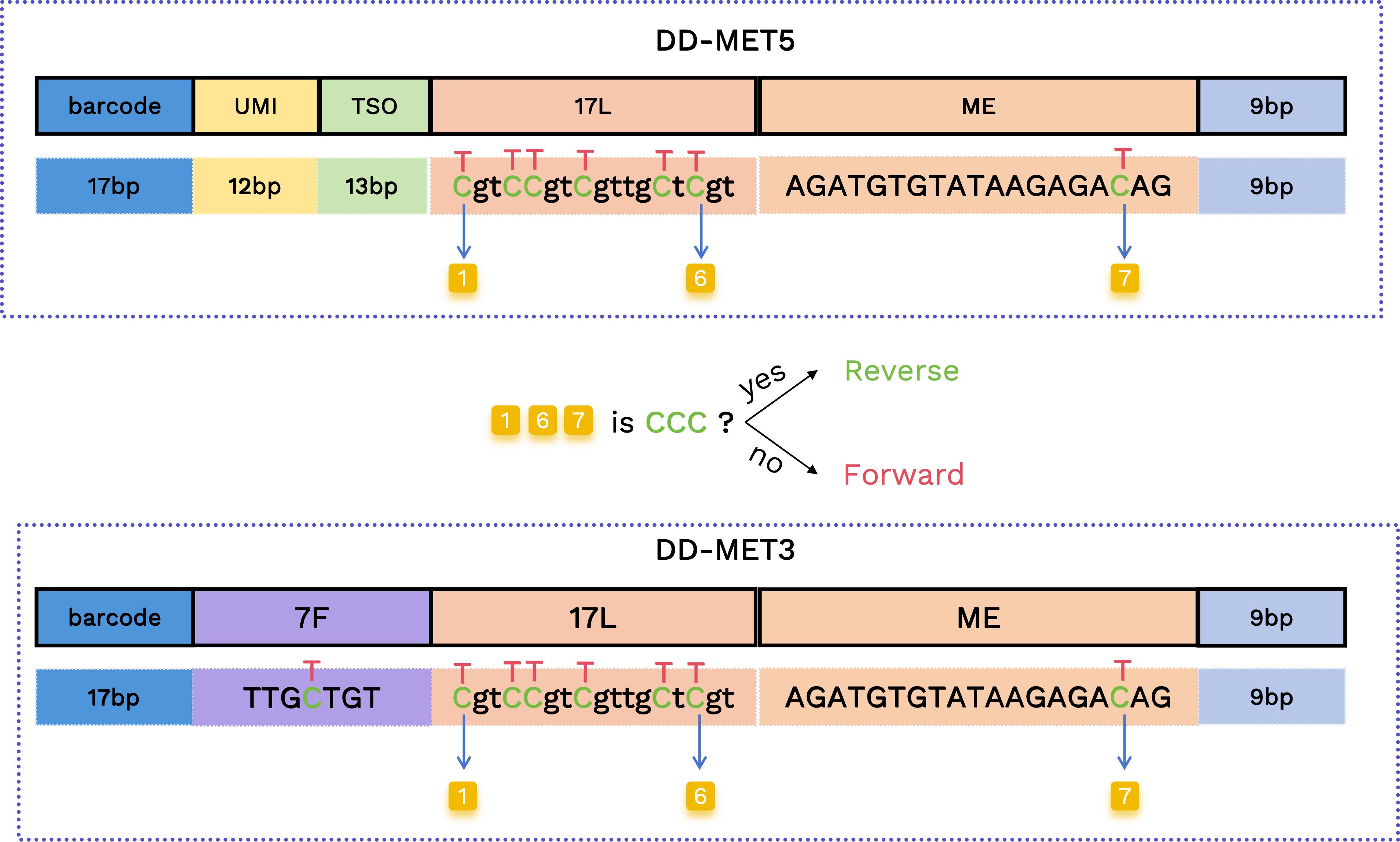
正反向 Reads 判定
从对应 17L 和 ME 的位置中,有 7 个碱基可以是 C 或转化后的 T(如下所示)。我们使用第一个和最后两个 C/T 位置来判定正向与反向 Reads:如果所有三个位置都是 C,则表示反向 Read;否则,为正向 Read。
正向: TgtTTgtTgttgTtTgtAGATGTGTATAAGAGAT
反向: CgtCCgtCgttgCtCgtAGATGTGTATAAGAGAC
在甲基化术语中,反向 Reads 对应 CTOT/CTOB(原始链的反向互补);正向 Reads 对应 OT/OB(原始链)。 “正向”或“反向”的判定会注释在 Read 名称中。
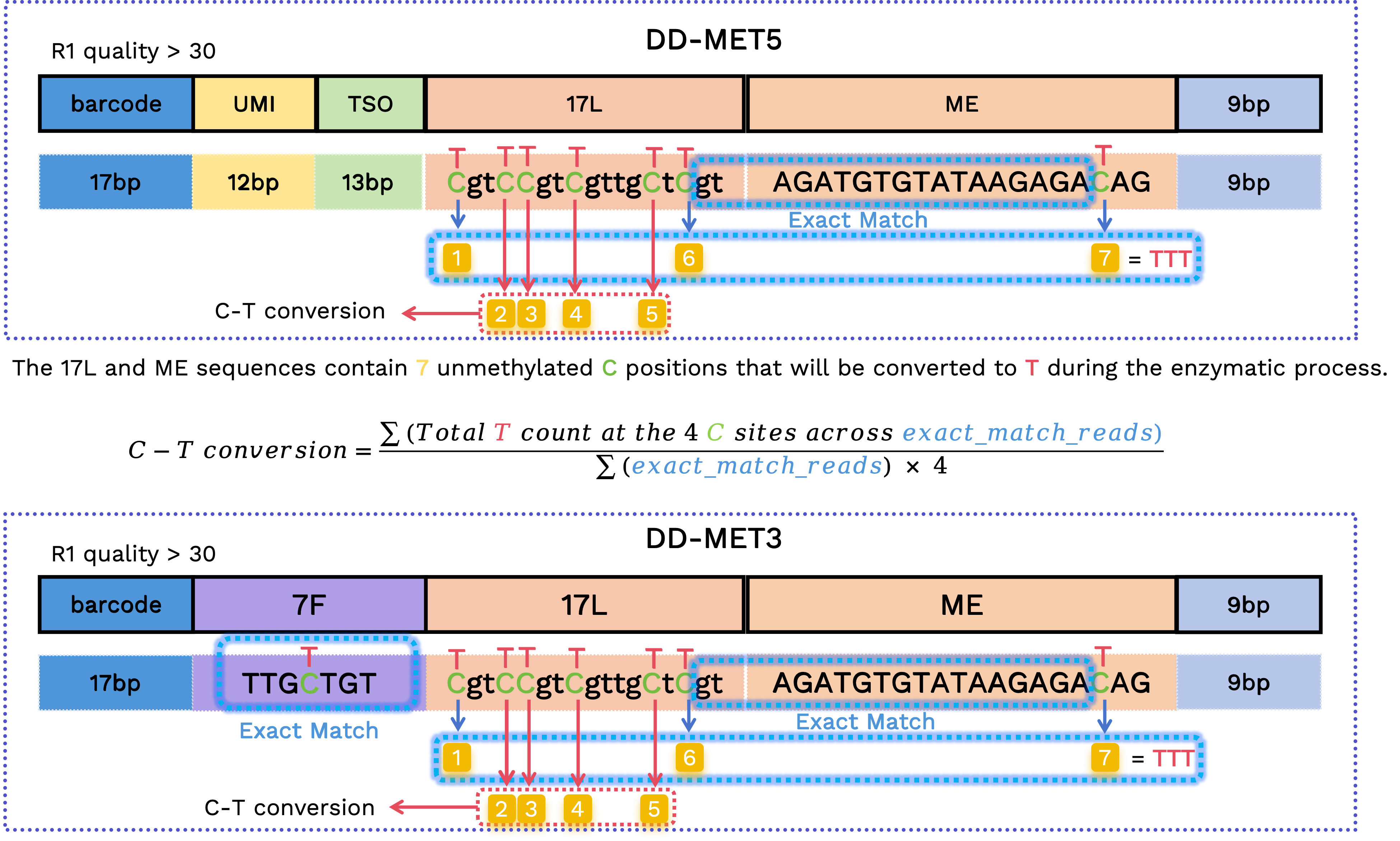
C-T 转化率
我们利用 17L 和 ME 序列中的原始 C 位置计算 C 到 T 的转化率。由于这些是固定的序列,容易受到测序错误的影响,我们仅使用结构经过验证的 reads 进行计算:
在 DD-MET3 中,7F 序列必须是
TTGCTGT或TTGTTGT,跨越 17L 和 ME 的序列必须是GTAGATGTGTATAAGAGA,并且第一个和最后两个原始 C 位置的碱基必须是 T。在 DD-MET5 中,跨越 17L 和 ME 的序列必须是
GTAGATGTGTATAAGAGA,并且第一个和最后两个原始 C 位置的碱基必须是 T。
对于保留的 reads,我们提取相应位置的碱基来计算 C 到 T 的转化率:
NOTE
上述过滤步骤仅用于计算 C 到 T 的转化率;不符合这些标准的 reads 不会从最终输出的 FASTQ 文件中过滤掉。
去除伪影
根据文库设计中的预定义位置,从 Read1 中去除 TSO/7F 接头、17L 和 ME 序列。
去接头
使用 cutadapt 去除由 R1/R2 通读事件(重叠的双端 reads)引入的 ME 接头序列。
去除 Tn5 转座酶引入的 9 bp 间隙
去除接头和其他人工序列后,我们额外去除 Tn5 转座引入的插入片段两侧的 9 bp 间隙。这 9 bp 区域可能携带人工甲基化信号,并虚假地提高插入边界附近的 CH 甲基化水平,因此在下游分析之前将其去除。
过滤非 CpG 甲基化 C 碱基过多的 Reads(可选)
根据 Read 对中非 CpG 甲基化 C 碱基的数量进行过滤。默认情况下,Read1/Read2 中检测到超过 2 个非 CpG 甲基化 C 的 Read 对将被去除。如果您不想启用此过滤器,请将 filter_ch 设置为 0。
NOTE
此过滤策略基于 Lu 等人 [1] 的发现,即合成接头中的切口可能触发 Bst 聚合酶的切口平移。这种活性会掺入 5-甲基-dCTP,导致出现完全未转化的 Reads,表现为人工甲基化信号。
过滤过短 Reads 经过上述过滤和去接头步骤后,如果 Read 对中 R1 的长度小于 20 bp 或 R2 的长度小于 60 bp,则该 Read 对将被过滤掉。
步骤 2:Bismark 比对和按名称排序
比对和标记
我们使用 Bismark 进行甲基化比对。我们修改后的 Bismark 增加了 --add_barcode 和 --add_umi 参数,以使用 CB(校正后的条形码)和 UR(原始 UMI)对 BAM 文件进行按 read 名称标记。对于正向 reads,我们使用 -X 1000 允许插入片段大小最大为 1000 bp;对于反向 reads,我们使用 --pbat 和 -X 1000。 比对后,使用 samtools sort -n 按 read 名称对 BAM 进行排序;按名称排序的 BAM 将作为下游分析的输入。
步骤 3:ALLCools 分析
按细胞条形码拆分
将按名称排序的 BAM 按转录组来源的细胞条形码拆分为单细胞 BAM 文件,每个文件包含一个细胞的唯一比对 reads。
生成 ALLC 文件
将每个单细胞 BAM 按位置排序,并使用 ALLCools bam-to-allc 转换为 ALLC。我们修改后的 ALLCools 执行基于 UR 标签的 UMI 校正和每个 C 位点的去重。
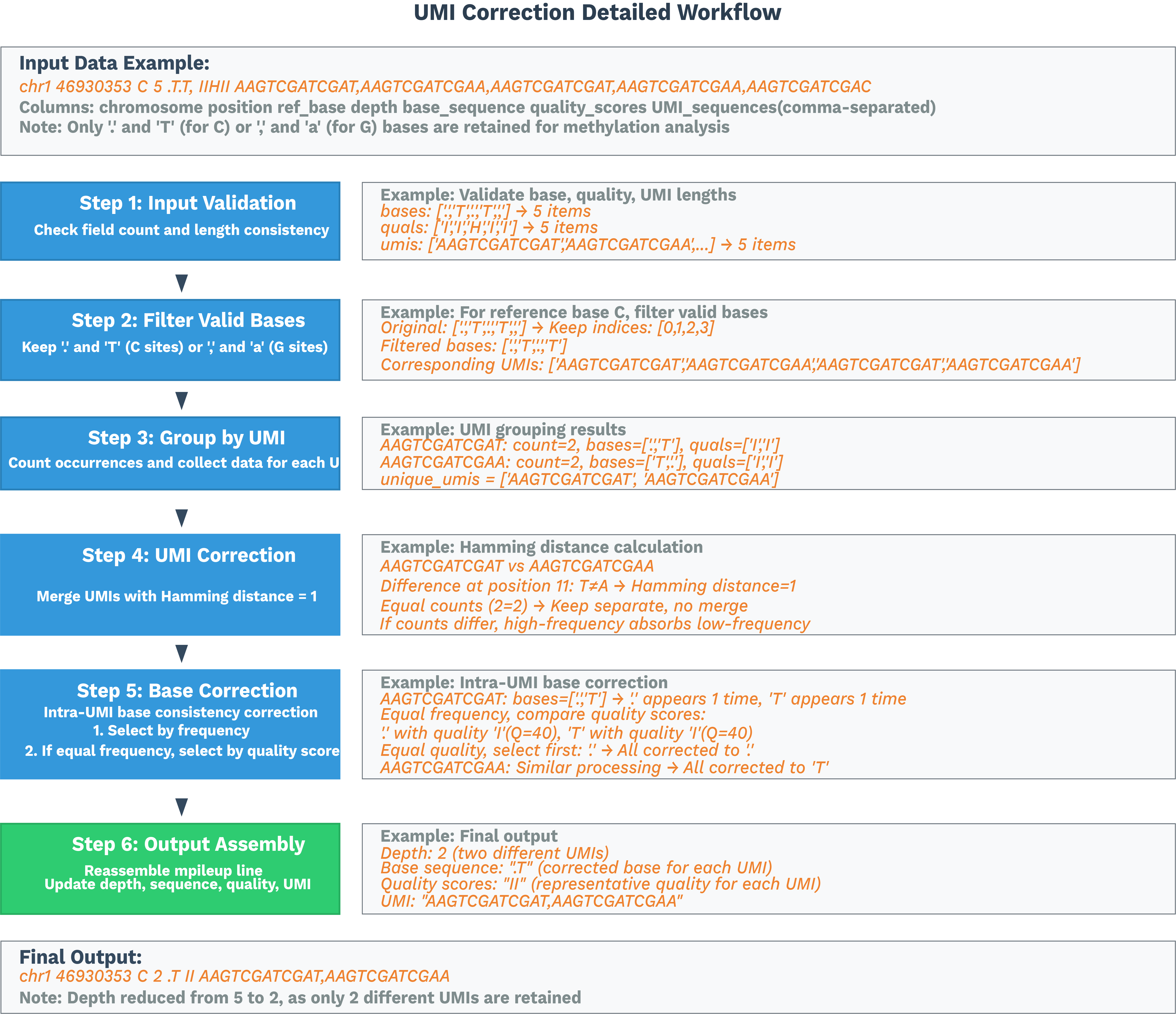
有关详细信息,请参阅 SeekGene ALLCools。
生成 MCDS
运行 allcools generate-dataset 对基因组进行分箱(chrom10k/20k/50k/100k/500k/1M/geneslop2k)并计算单细胞甲基化矩阵。Geneslop2k bin 定义为每个基因两侧各 2k bp。
步骤 4:降维和聚类
默认情况下,使用 ALLCools 对 chrom20k bin 执行 LSI 降维,然后进行 UMAP 可视化。